Chemia koloidalna. Chemia fizyczna i koloidalna. Zjawiska powierzchniowe i adsorpcja
Przeczytaj także
Plan:
Systemy rozproszone.
Struktura miceli koloidalnej.
Metody otrzymywania koloidów liofobowych (CL).
Temat i znaczenie chemia koloidów.
Chemia koloidów- Ten nauka o układach rozproszonych i zjawiskach powierzchniowych powstających na granicy faz.
Chemia koloidalna jest chemia ciał rzeczywistych, ponieważ rzeczywiste obiekty są żywe i przyroda nieożywiona produkty i materiały tworzone i wykorzystywane przez człowieka niemal zawsze występują w stanie rozproszonym, tj. zawierają drobne cząstki, cienkie filmy, membrany, włókna z jasno określonymi granicami. Jednocześnie zjawiska powierzchniowe i układy rozproszone zachodzą daleko poza Ziemią. Na przykład materią międzygwiazdową są obłoki gazu i pyłu. Zjawiska meteorologiczne - burze, deszcz, śnieg, grad, mgła i inne - są procesami koloidalnymi.
Chemia koloidów stanowi podstawę naukową produkcja tworzyw sztucznych, gumy, włókien syntetycznych, klejów, farb i materiały budowlane, żywność, lekarstwa itp. Praktycznie nie ma dziedziny przemysłu, która w takim czy innym stopniu nie zajmuje się układami koloidalnymi.
Rola chemii koloidalnej jest również ogromna w rozwiązywaniu szeregu problemów konserwatorskich. środowisko włączając sprzątanie Ścieki, uzdatnianie wody, wychwytywanie aerozoli, kontrola erozji gleby itp.
Chemia koloidów otwiera nowe podejścia badać historię skorupy ziemskiej, ustalać powiązania między właściwościami koloidowo-chemicznymi gleby a jej żyznością, wyjaśniać warunki powstania życia, mechanizmy życia; ona jest jedną z wiodących fundacji współczesna biologia, gleboznawstwo, geologia, meteorologia. Razem z biochemią i chemią fizyczną polimerów stanowi podstawy doktryny o pochodzeniu i rozwoju życia na Ziemi. Fakt, że wszystkie żywe układy są bardzo rozproszone, podkreśla znaczenie chemii koloidalnej dla rozwoju całej współczesnej nauki.
Znaczenie procesów koloidalnych w rolnictwie jest ogromne (tworzenie dymu i mgły do zwalczania szkodników rolniczych, granulacja nawozów, poprawa struktury gleby itp.). Procesy kulinarne: dojrzewanie galaretek (czerwienienie chleba, oddzielanie się płynu od galaretek, galaretek itp.), adsorpcja (klarowanie bulionów) to procesy koloidalne, które leżą u podstaw pieczenia, winiarstwa, warzenia piwa i innej produkcji żywności.
2. Układy rozproszone.
Systemy rozproszone- są to układy, w których występuje jedna substancja w postaci cząstek różne rozmiary rozproszone w innej substancji.
W układach dyspersyjnych rozróżnia się fazę rozproszoną (DP) - drobno rozdrobnioną substancję i ośrodek dyspersyjny (DS) - substancję jednorodną, w której rozprowadzona jest faza rozproszona (w mętna woda zawierające glinę, DF to stałe cząstki gliny, a DS to woda).
Ważną cechą systemów dyspersyjnych jest stopień dyspersji - średni rozmiar cząstki fazy rozproszonej.
Ze względu na stopień dyspersji zazwyczaj wyróżnia się następujące klasy układów rozproszonych:
Grube systemy– układy, w których wielkość cząstek fazy rozproszonej przekracza 10 -7 m (zawiesiny i emulsje).
Układy koloidalne– układy, w których wielkość cząstek fazy rozproszonej wynosi 10 -7 – 10 -9 m. Są to układy mikroheterogeniczne z dobrze rozwiniętą granicą międzyfazową. Ich cząsteczki nie osiadają pod wpływem grawitacji i nie przechodzą przez filtry papierowe, lecz są zatrzymywane przez błony roślinne i zwierzęce. Na przykład roztwory białek, koloidy glebowe itp.
Czasami izolowane są molekularne (jonowe) układy rozproszone, które, ściśle rzecz biorąc, są prawdziwe rozwiązania, tj. układy jednorodne, ponieważ nie mają granic faz. Wielkość cząstek fazy rozproszonej jest mniejsza niż 10 -9 m. Rozpuszczona substancja ma postać cząsteczek lub jonów. Na przykład roztwory elektrolitów, cukru.
Z kolei układy koloidalne dzielą się na dwie grupy, znacznie różniące się charakterem oddziaływań między cząstkami fazy rozproszonej a ośrodkiem dyspersyjnym - liofobowe roztwory koloidalne (zole) oraz roztwory związków o dużej masie cząsteczkowej (HMC), które wcześniej nazywano koloidy liofilowe.
DO koloidy liofobowe Należą do nich układy, w których cząstki fazy rozproszonej słabo oddziałują z ośrodkiem dyspersyjnym; układy te można otrzymać jedynie przy wydatku energii i są stabilne jedynie w obecności stabilizatorów.
Rozwiązania wkładki wewnątrzmacicznej powstają samoistnie w wyniku silnego oddziaływania cząstek fazy rozproszonej z ośrodkiem dyspersyjnym i mogą pozostać stabilne bez stabilizatorów.
Koloidy liofobowe i roztwory domaciczne różnią się składem fazy rozproszonej. Do koloidów liofobowych jednostka struktury jest złożonym, wieloskładnikowym agregatem o zmiennym składzie – micela, dla rozwiązań IUD – makrocząsteczka.
Układy rozproszone dzielą się na grupy różniące się charakterem i stanem agregacji fazy rozproszonej i ośrodka dyspersyjnego:
Jeśli ośrodek dyspersyjny jest ciekły, a fazą rozproszoną są cząstki stałe, układ nazywa się zawiesiną lub zawieszenie;
Jeżeli faza rozproszona składa się z kropelek cieczy, wówczas układ nazywa się emulsja. Z kolei emulsje dzielą się na dwa typy: prosty, Lub „olej w wodzie”(kiedy fazą rozproszoną jest ciecz niepolarna, a ośrodkiem dyspersyjnym jest ciecz polarna) i odwracać, Lub „woda w oleju”(kiedy ciecz polarna jest rozproszona w cieczy niepolarnej).
Do systemów rozproszonych zalicza się również systemy rozproszone piana(gaz rozproszony w cieczy) i ciała porowate(faza stała, w której rozproszony jest gaz lub ciecz). Główne typy systemów dyspersyjnych podano w tabeli.
3.Struktura miceli koloidalnej.
Cząstki DF w koloidach liofobowych mają złożoną strukturę, zależną od składu DF, DS i warunków otrzymania roztworu koloidalnego. Warunkiem koniecznym otrzymania stabilnych zoli jest obecność trzeciego składnika, który pełni rolę stabilizatora.
Zdyspergowana cząstka – micela składa się z:
jądra w stanie krystalicznym lub ciekłym;
jednocząsteczkowa warstwa adsorpcyjna Jony determinujące potencjał;
płynna powłoka, bardziej zagęszczona na powierzchni cząstki i stopniowo zamieniająca się w zwykły ośrodek dyspersyjny;
mocno związany warstwa przeciwjonów, tj. jony niosące ładunek przeciwny do znaku ładunku jonów determinujących potencjał;
warstwa dyfuzyjna przeciwjony, które poruszają się swobodnie podczas elektroforezy lub elektroosmozy.
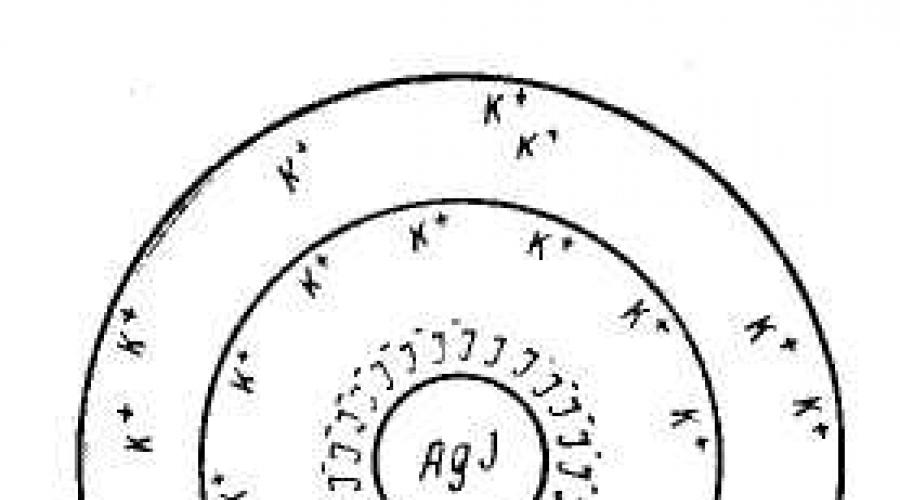
Struktura jednostka strukturalna koloidy liofobowe – micele– można pokazać jedynie schematycznie, gdyż micela nie ma określonego składu. Rozważmy strukturę miceli koloidalnej na przykładzie hydrozol jodku srebra, otrzymany w wyniku reakcji rozcieńczonych roztworów azotanu srebra i jodku potasu:
AgNO 3 + KI ––> AgI + KNO 3
Koloidalna micela zolu jodku srebra jest utworzona przez mikrokryształ AgI, który ma zdolność selektywnej adsorpcji kationów Ag + lub I - ze środowiska. Aby uzyskać stabilny zol, konieczna jest obecność jednego z elektrolitów AgNO 3 lub KI w nadmiarze jako stabilizatora.
Jeżeli reakcję prowadzi się w nadmiarze jodku potasu, kryształ zaadsorbuje I -; przy nadmiarze azotanu srebra mikrokryształ adsorbuje jony Ag+. W rezultacie mikrokryształ uzyskuje ładunek ujemny lub dodatni.
1. Nadmiar KI
Nierozpuszczalne cząsteczki AgI formularz rdzeń cząstki koloidalnej (miceli) M[ AgI].
I - jony są adsorbowane na powierzchni rdzenia (zwykle te jony, które są częścią rdzenia, tj. Są adsorbowane w tym przypadku Ag + lub I -), nadając mu ładunek ujemny. Uzupełniają sieć krystaliczną jądra, mocno wchodząc w jego strukturę, tworząc warstwa adsorpcyjna M[ AgI] · nI – . Nazywa się jony, które są zaadsorbowane na powierzchni jądra i nadają mu odpowiedni ładunek Jony determinujące potencjał.
Zaadsorbowane jony określające potencjał przyciągają jony z roztworu przeciwny znak przeciwjony(K+), a część z nich (n-x) jest adsorbowana na cząstce { M[ AgI] · nI – · (N- X) K + } X – . Rdzeń + warstwa adsorpcyjna = granulka.
Powstają pozostałe przeciwjony rozproszona warstwa jonów.
Reprezentuje rdzeń z warstwami adsorpcyjnymi i rozproszonymi micela.
Otrzymany schematyczny diagram miceli zolu jodku srebra nadmiar jodku potasu (jonami determinującymi potencjał są I – aniony, przeciwjony to jony K+) można przedstawić w następujący sposób:
(m · nI – · (n-x)K + ) x– · xK +

2. Po otrzymaniu zolu jodku srebra w nadmiarze azotanu srebra cząstki koloidalne będą miały ładunek dodatni:
(m nAg + (n-x)NO 3 – ) x+ x NO 3 –


S. V. Egorov, E. S. Orobeyko, E. S. Mukhacheva
Chemia koloidalna, ściągawka
1. Powstanie i główne etapy rozwoju chemii koloidalnej. Przedmiot i przedmioty badań chemii koloidalnej
Powstanie nauki chemii koloidalnej wiąże się z badaniami angielskiego chemika T. Grahama . Po pionierskich badaniach M. Faradaya (1857), kiedy po raz pierwszy otrzymano stabilne roztwory koloidalne wysoko zdyspergowanego złota, w 1861 Graham badał dyfuzję różnych substancji w roztworach wodnych i odkrył, że niektóre z nich (żelatyna, agar-agar itp.) dyfuzują w wodzie znacznie wolniej niż na przykład sole i kwasy. Również w przypadku przesycenia roztworów substancje te nie krystalizowały, lecz tworzyły galaretowatą, lepką masę. T. Graham nazwał te substancje koloidami (od greckiego kolla – „klej”, eidos – „rodzaj”). Tak pojawiła się nazwa nauki - „chemia koloidów”. T. Graham wysunął hipotezę o istnieniu w przyrodzie dwóch przeciwstawnych klas substancje chemiczne– krystaloidy i koloidy. Pomysł ten zainteresował wielu naukowców i w drugiej połowie XIX wieku. Rozpoczął się szybki rozwój chemii koloidów. W Rosji w tym czasie dużą uwagę poświęcono chemii koloidów, głównie pod wpływem DI Mendelejew . Badania zależność od temperatury napięcie powierzchniowe cieczy organicznych (1861) doprowadził Mendelejewa do odkrycia tej koncepcji krytyczna temperatura Substancje. Mendelejew wyraził także ideę związku między napięciem powierzchniowym a innymi właściwościami materii. W ciągu tych lat odkryto wiele substancji o właściwościach koloidalnych, m.in. różne metody oczyszczania i stabilizacji koloidów, stworzono metody ich badania. Wraz z odkryciem nowych koloidów w pierwszej połowie XX wieku hipoteza T. Grahama została zastąpiona. przyszedł do koncepcja powszechności koloidalnego (rozproszonego) stanu skupienia:„Stan koloidalny nie jest determinowany składem substancji. W pewnych warunkach każda substancja może znajdować się w stanie koloidalnym.” Koncepcję tę sformułował profesor Instytutu Górnictwa w Petersburgu P. P. Weymarn V 1906–1910. Pokazał, że typowe koloidy (np. żelatynę) można wyizolować w postaci krystalicznej i odwrotnie, z substancji krystaloidalnych (np. sól kuchenna w benzenie). Nastąpiła zmiana priorytetów chemii koloidalnej. Głównym kierunkiem było badanie stanu rozproszonego (koloidalnego) substancji. Około lat dwudziestych XX wieku. podstawowe problemy chemii koloidalnej umownie dzieli się na trzy grupy: skład, struktura i właściwości cząstek koloidalnych; oddziaływanie cząstek z ośrodkiem rozproszonym; oddziaływania kontaktowe cząstek ze sobą, prowadzące do powstania struktur koloidalnych. W tym okresie odkryto podstawowe prawa chemii koloidalnej - prawo ruchu Browna i dyfuzję cząstek koloidalnych (A.Einstein) , heterogeniczny charakter roztworów koloidalnych (R. Zsigmondy) , równowaga sedymentacyjno-dyfuzyjna dyspersji w polu grawitacyjnym (J.Perrin) i w wirówce (T. Svedberg) , rozpraszanie światła (J. Rayleigh) , koagulacja zoli z elektrolitami (G. Schulze I V. Hardy) . Pojawienie się w drugiej połowie XX wieku. wysokorozdzielcze metody badania struktury substancji (NMR, mikroskopia elektronowa i sił atomowych, modelowanie komputerowe, spektroskopia korelacji fotonów itp.) umożliwiły przejście do systematycznych badań struktury i właściwości układów koloidalnych. Współczesna definicja tej nauki brzmi: chemia koloidów to doktryna o właściwościach i przemianach substancji w stanach rozproszonych i ultradyspersyjnych oraz zjawiskach powierzchniowych w układach rozproszonych. Obiekty badań w chemii koloidalnej mają wysoko rozwiniętą powierzchnię i reprezentują różne zole, zawiesiny, emulsje, pianki, filmy powierzchniowe, membrany i ciała porowate, układy nanostrukturalne (nanorurki, filmy Langmuira-Blodgetta, hybrydowe organiczne i nieorganiczne materiały kompozytowe, nanokompozyty).
2. Główne cechy systemów rozproszonych. Cechy stanu ultramikroheterogenicznego (nanostanu)
Systemy rozproszone utworzone z dwóch lub więcej faz z wysoce rozwiniętą granicą międzyfazową, przy czym co najmniej jedna z faz jest faza rozproszona– rozmieszczone w postaci drobnych cząstek (kryształów, kropel, pęcherzyków itp.) w innej, ciągłej fazie – ośrodek dyspersyjny. Przykładami są skały, grunty, gleby, dym, chmury, opady atmosferyczne, tkanki roślinne i zwierzęce itp. Najważniejszą cechą systemów rozproszonych jest niejednorodność. Funkcja systemy rozproszone– silnie rozwinięta powierzchnia międzyfazowa i co za tym idzie duża energia swobodna, dlatego układy zazwyczaj rozproszone (z wyjątkiem liofilowych) są termodynamicznie niestabilne. Mają zwiększoną zdolność adsorpcji, aktywność chemiczną, a czasem biologiczną. Układy rozproszone charakteryzują się wzrostem pola powierzchni wraz ze wzrostem dyspersji i rosnącą rolą zjawisk powierzchniowych. Układy rozproszone charakteryzują się bardzo dużą powierzchnią właściwą W faza rozproszona.
W < K/dr,
Gdzie K– współczynnik bezwymiarowy (dla cząstek kulistych i sześciennych K = 6); R– gęstość fazy rozproszonej.
Innymi ważnymi parametrami termodynamicznymi charakteryzującymi układy koloidalne są specyficzna swobodna energia powierzchniowa σ (napięcie powierzchniowe), entropia powierzchniowa H i specyficzna adsorpcja G. Ważna funkcja układach rozproszonych polega na tym, że znaczna część całkowitej masy i energii swobodnej układu jest skoncentrowana w międzyfazowych warstwach powierzchniowych. Z tą funkcją powiązane są następujące właściwości: niepowtarzalność(Lub indywidualność) systemy ze względu na nierówną powierzchnię cząstek fazy rozproszonej, które mają różne energie powierzchniowe nawet przy tej samej powierzchni właściwej; strukturyzacja, związany z tendencją do niestabilności termodynamicznej. Podstawowa właściwość układy rozproszone to ich zdolność do stopniowej ewolucji, co jest związane z naturą rozproszonego stanu materii, przede wszystkim z nierównowagą termodynamiczną. Nadmiar energii swobodnej spowodowany obecnością wysoko rozwiniętej granicy faz pomiędzy fazą rozproszoną a ośrodkiem dyspersyjnym stymuluje przepływ różne procesy(fizyczne, fizykochemiczne), co prowadzi do zmniejszenia energii swobodnej Helmholtza F. Znak jak labilność, jest konsekwencją niestabilności termodynamicznej i tendencji do zmniejszania energii swobodnej poprzez tworzenie mniej rozproszonych struktur. Główna charakterystyka układy rozproszone - rozmiary cząstek (lub dyspersja), który jest określony przez stosunek Całkowita powierzchnia powierzchni międzyfazowej do objętości fazy rozproszonej. W oparciu o to kryterium rozróżnia się grubo (słabo rozproszone) (cząstki mają wielkość 10–4 cm i więcej) i drobno rozproszone (silnie rozproszone) (cząstki mają wielkość 10–4 do 10–5–10–7 cm), lub układy koloidalne (koloidy). Graniczny stopień dyspersji, przy którym układ koloidalny zachowuje swoją główną właściwość – niejednorodność – mieści się w zakresie od 1 do 100 nm. Zajmują najdrobniejsze cząsteczki pozycja pośrednia pomiędzy cząsteczkami (atomami, jonami) a ciałami makroskopowymi (fazami). Rozmiar cząstek fazy rozproszonej D jest zbliżony do maksymalnego, tym silniejszy będzie wpływ efektu skali – zależności właściwości od wielkości cząstek. Jeżeli dla układów o średnim stopniu dyspersji wyznacza się jedynie napięcie powierzchniowe s skład chemiczny, to w przypadku nanosystemów konieczne jest już uwzględnienie zależności napięcia powierzchniowego od wielkości rozproszonych cząstek.
3. Różne rodzaje klasyfikacja układów rozproszonych. Liofilowe i liofobowe systemy dyspersyjne
Systemy rozproszone heterogeniczny i składa się z dwóch faz, z których jedna jest jedna (faza rozproszona) w postaci cząstek różnej wielkości rozmieszczonych w innej fazie – ciągłej ośrodek dyspersyjny. Układy rozproszone klasyfikuje się przede wszystkim według wielkości cząstek fazy rozproszonej (lub stopnia dyspersji). Ponadto dzieli się je na grupy, które różnią się charakterem i stanem agregacji fazy rozproszonej i ośrodka dyspersyjnego (może być stały, ciekły i gazowy), budową i charakterem oddziaływań międzyfazowych. Jeśli ośrodek dyspersyjny jest ciekły, a fazą rozproszoną są cząstki stałe, układ nazywa się zawiesiną lub zawiesiną; jeżeli faza rozproszona składa się z kropelek cieczy, wówczas układ nazywa się emulsją. Do układów rozproszonych zalicza się także pianki (gaz rozproszony w cieczy), aerozole (ciecz w gazie) oraz ciała porowate (faza stała, w której rozproszony jest gaz lub ciecz). Skrócony typ układu dyspersyjnego w zależności od stan skupienia zapisywana jako ułamek, gdzie w liczniku znajduje się faza rozproszona, a w mianowniku ośrodek dyspersyjny (np. T/T (stałe roztwory koloidalne – minerały, stopy), T/L (zole – zawiesiny), T/G (aerozole - pyły, opary); L/T (ciała porowate - żele), L/L (emulsje), L/G (aerozole - mgły); G/T (układy porowate i kapilarne), G/L (pianki - emulsje gazowe)). Systemy H/G zwykle nie pojawiają się w klasyfikacji, ponieważ... warunek konieczny tworzenie układu dyspersyjnego – ograniczona rozpuszczalność substancji w ośrodku.
Chemia koloidalna to nauka o właściwościach fizycznych i chemicznych układów rozproszonych oraz zjawisk powierzchniowych.
Układ rozproszony (DS) to układ, w którym co najmniej jedna substancja w stanie mniej lub bardziej rozdrobnionym (zdyspergowanym) jest równomiernie rozłożona w masie innej substancji. DS jest heterogeniczny; składa się z co najmniej dwóch faz. Fazę rozdrobnioną nazywa się fazą rozproszoną. Ośrodek ciągły, w którym faza rozproszona jest fragmentowana, nazywany jest ośrodkiem dyspersyjnym. Cechą charakterystyczną DS jest obecność dużej powierzchni międzyfazowej. Pod tym względem właściwościami decydującymi są właściwości powierzchni, a nie cząstek jako całości. DS charakteryzuje się procesami zachodzącymi na powierzchni, a nie wewnątrz fazy.
Zjawiska powierzchniowe i adsorpcja
Zjawiska powierzchniowe to zjawiska zachodzące na granicy faz układów rozproszonych. Należą do nich: napięcie powierzchniowe, zwilżanie, adsorpcja itp. Najważniejsze procesy techniczne: oczyszczanie powietrza i ścieków ze szkodliwych zanieczyszczeń, wzbogacanie rud mineralnych (flotacja), spawanie metali, czyszczenie, smarowanie, malowanie różnych powierzchni i wiele innych.
Napięcie powierzchniowe
Każdy interfejs fazowy ma specjalne właściwości, które różnią się od właściwości wewnętrznych części sąsiednich faz. Dzieje się tak dlatego, że warstwy powierzchniowe posiadają nadmiar energii swobodnej. Rozważmy układ składający się z cieczy i gazu (ryc. 1).
Na cząsteczkę A, znajdujący się wewnątrz cieczy, siły wzajemnego przyciągania działają na część wszystkich otaczających go sąsiadujących cząsteczek. Wynik tych sił wynosi zero. Dla cząsteczki W, znajdujące się na powierzchni cieczy, nie wszystkie siły przyciągania molekularnego zostaną skompensowane. Wynika to z faktu, że w gazie cząsteczki są daleko od siebie, a siły przyciągania między nimi są znikome. Dlatego cząsteczki W doświadczaj przyciągania wyłącznie przez ciecz. Dla nich wypadkowa sił przyciągania molekularnego nie jest zerowa i jest skierowana w głąb fazy ciekłej. Ta siła nazywa się Ciśnienie wewnętrzne. Ciśnienie to ma tendencję do wciągania wszystkich cząsteczek z powierzchni w głąb cieczy. Pod tym ciśnieniem ciecz kurczy się i zachowuje, jakby miała „skórkę”. Im bardziej różnią się interakcje międzycząsteczkowe w sąsiednich fazach, tym większe jest ciśnienie wewnętrzne.
W celu utworzenia nowa powierzchnia separacja faz, na przykład w celu rozciągnięcia cieczy w folię, konieczne jest wykonanie pracy przeciw wewnętrznym siłom ciśnienia. Im większe ciśnienie wewnętrzne, tym więcej energii potrzeba. Energia ta skupia się w cząsteczkach znajdujących się na powierzchni i nazywa się ją darmowa energia powierzchniowa.
Nazywa się pracę wykonaną na utworzenie 1 cm 2 granicy faz lub jej równoważną swobodną energię powierzchniową napięcie powierzchniowe i oznaczać , J/m2. Wtedy podaż darmowej energii (F s) skupionej na granicy faz (S) jest równa: F s = S. Dlatego niż mniejszy rozmiar cząstek, tym większa jest powierzchnia S i tym większa rezerwa wolnej energii powierzchniowej ten rozproszony system ma w porównaniu z konwencjonalnymi masywnymi ciałami.
Z termodynamiki wiadomo, że warunek stabilnej równowagi układu to minimalna energia swobodna. Pod tym względem systemy dyspersyjne są niestabilne termodynamicznie: w nich procesy zachodzą samoistnie , związany ze zmniejszeniem granicy faz w wyniku powiększenia cząstek. Oczywiście stan równowagi odpowiada stratyfikacja systemu (na przykład emulsja dzieli się na dwie ciecze, a zawiesina na ciecz i osad). Ponadto, ponieważ wartość dąży do minimum, przyjmuje ciecz w stanie wolnym kształt kuli, (krople płynu). Wyjaśnia to fakt, że powierzchnia kuli jest minimalna dla danej objętości materii.
Minimalną wartość Fs, czyli stan równowagi układu, można również osiągnąć dążąc do wartości minimalnej . Zatem, spontaniczny w układach rozproszonych zachodzą także procesy związane ze spadkiem napięcia powierzchniowego. Dla ciała stałe , które nie mogą zmienić swojego kształtu tak łatwo jak ciecze, swobodna energia powierzchniowa F s może się zmniejszyć tylko jedna droga ─ wskutek spadku napięcia powierzchniowego . Dzieje się tak: cząsteczki leżą warstwa powierzchniowa, są w stanie przyciągać, a czasami bardzo mocno trzymać inne cząsteczki ze środowiska otaczającego ciało stałe. Zjawisko to nazywa się sorpcja.
Na wartość napięcia powierzchniowego wpływają:
1. Charakter substancji . Ogrom zależy od struktury fazy skondensowanej, czyli charakteru sił działających pomiędzy cząsteczkami. Im większa polarność wiązań chemicznych w substancji, tym wyższe wartości charakterystyczny dla tej substancji. Wśród cieczy (na granicy z powietrzem) największą wartość ma woda. Jeszcze wyższe wartości obserwowane w stopionych kryształach jonowych i metalach stałych.
2.Temperatura. Wraz ze wzrostem temperatury wartość maleje, ponieważ ruch termiczny cząstki po podgrzaniu osłabiają działanie sił międzycząstkowych w substancji.
3.Stężenia dodanych dodatków. Ogrom zależy od stężenia substancji rozpuszczonych w cieczy testowej. Istnieją dwa rodzaje substancji. Powierzchnia ─ substancje nieaktywne (PIS), wychowywanie napięcie powierzchniowe roztworu w porównaniu z czystym rozpuszczalnikiem. Należą do nich większość mocnych elektrolitów.
Środki powierzchniowo czynne (środek powierzchniowo czynny), silnie obniżenie poziomu napięcie powierzchniowe powstałego roztworu. Wraz ze wzrostem stężenia środka powierzchniowo czynnego w roztworze wartość gwałtownie maleje, ponieważ substancja jest skoncentrowana (sorbowana) w powierzchniowej warstwie roztworu i nie jest równomiernie rozłożona w całej objętości roztworu. W roztworach wodnych polarne związki organiczne wykazują aktywność powierzchniową - alkohole, kwasy, sole itp. Cząsteczki takich związków zawierają jednocześnie grupy polarne (O, OH, COOH, NH2) i niepolarny łańcuch węglowodorowy. Schematycznie cząsteczka środka powierzchniowo czynnego jest konwencjonalnie oznaczona w następujący sposób: „O────”. Typowym przykładem środka powierzchniowo czynnego jest sól sodowa kwasu stearynowego C17H35COONa (mydło w postaci stałej).
Przedmiot chemii koloidów
Układy koloidalne i przedmiot chemii koloidalnej
Układy koloidalne
Początkowo chemia koloidalna była tylko rozdziałem Chemia fizyczna. Obecnie jest to niezależna dyscyplina posiadająca własny wachlarz pomysłów. Opracowano specjalne metody badań koloidowo-chemicznych: ultramikroskopię, mikroskopia elektronowa, ultrawirowanie, elektroforeza itp. Praktyka pokazała ogromne znaczenie chemii koloidalnej dla nowoczesna technologia. Nie można określić branży Gospodarka narodowa, w których nie byłyby stosowane układy koloidalne i procesy koloidalne. Człowiek od niepamiętnych czasów miał do czynienia z układami koloidalnymi. Jednak ich badania rozpoczęły się stosunkowo niedawno.
Powszechnie uważa się, że twórcą chemii koloidów jest angielski naukowiec Thomas Graham (*) (1805-1869), który w latach 50-60 ubiegłego wieku wprowadził do obiegu podstawowe pojęcia chemii koloidów. Nie zapominajmy jednak, że miał on poprzedników, a przede wszystkim Jacoba Berzeliusa (*) i włoskiego chemika Francesco Selmiego (*). W latach 30. XIX w. Berzelius opisał szereg osadów, które podczas przemywania przechodzą przez filtr (kwasy krzemowy i wanadowy, chlorek srebra, błękit pruski itp.). Berzelius nazwał te osady przechodzące przez filtr „roztworami”, ale jednocześnie zwrócił uwagę na ich ścisłe powinowactwo z emulsjami i zawiesinami, których właściwości były mu dobrze znane. Francesco Selmi w latach 50. XIX wieku kontynuował prace w tym kierunku, poszukując różnic fizykochemicznych pomiędzy układami utworzonymi przez przechodzące przez filtr osady (nazwał je „pseudoroztworami”) a zwykłymi roztworami prawdziwymi.
Angielski naukowiec Michael Faraday (*) w 1857 roku zsyntetyzował koloidalne roztwory złota – zawiesinę Au w wodzie o wielkości cząstek od 1 do 10 nm. i opracowane metody ich stabilizacji.
Te „pseudoroztwory” rozpraszają światło, rozpuszczone w nich substancje wytrącają się po dodaniu niewielkich ilości soli, przejściu substancji do roztworu i wytrącaniu się z niego nie towarzyszy zmiana temperatury i objętości układu, co zwykle obserwuje się podczas rozpuszczania substancji krystalicznych.
Thomas Graham rozwinął te pomysły dotyczące różnicy między „pseudoroztworami” a roztworami prawdziwymi i wprowadził pojęcie „koloidu”. Graham odkrył, że substancje zdolne do tworzenia galaretowatych, amorficznych osadów, takie jak wodorotlenek glinu, albumina, żelatyna, dyfundują w wodzie z mniejszą prędkością w porównaniu do substancji krystalicznych (NaCl, sacharoza). Jednocześnie substancje krystaliczne z łatwością przechodzą przez powłoki pergaminowe w roztworze („dializują”), ale substancje galaretowate nie przechodzą przez te skorupy. Uznając klej za typowego przedstawiciela substancji galaretowatych, niedyfuzyjnych i niedialitycznych, Graham nadał im Nazwa zwyczajowa„koloid”, tj. podobny do kleju (od greckiego słowa kolla - klej). Substancje krystaliczne oraz substancje, które dobrze dyfundują i dializują, nazwał „krystaloidami”.
Wymieńmy anomalne właściwości niektórych roztworów, które obecnie nazywamy układami koloidalnymi.
Właściwości układów koloidalnych:
1. rozpraszanie światła (opalescencja) (wskazuje na niejednorodność, układ wielofazowy).
Opalescencja staje się szczególnie zauważalna, gdy tak jak Tyndall (*) przepuszcza się wiązkę zbiegających się promieni przez roztwór koloidalny, umieszczając soczewkę pomiędzy źródłem światła a kuwetą z roztworem. W tym przypadku rozwiązania przezroczyste w świetle przechodzącym, m.in oświetlenie boczne wykazują wszystkie właściwości mętnego podłoża. W cieczy koloidalnej widzianej z boku tworzy się jasny stożek świetlny (stożek Tyndalla).
2. powolna dyfuzja
3. niskie ciśnienie osmotyczne
(pozycje 2 i 3 wskazują na obecność dużych cząstek w układzie)
4. roztwory koloidalne nadają się do dializy, tj. można oddzielić od zanieczyszczeń za pomocą membrany
5. zdolny do koagulacji (zniszczenia) układu podczas: dodawania zanieczyszczeń, zmiany T, mieszania itp.
6. czasami odkrywają zjawisko elektroforezy, odkryte przez Reussa (6) w Rosji w 1808 r., tj. cząstki w układzie mogą mieć ładunek.
Aby wyobrazić sobie, czym zajmuje się nauka „chemii koloidalnej”, należy odpowiedzieć na pytanie, czym są koloidy lub układy koloidalne?
Przedmiot chemii koloidów
Chemia koloidów – nauka o zjawiskach powierzchniowych i układach rozproszonych.
DO zjawiska powierzchowne Należą do nich procesy zachodzące na granicy faz, w warstwie powierzchniowej międzyfazowej, oraz powstające w wyniku oddziaływania faz sprzężonych.
Przypomnijmy to faza jest częścią układu termodynamicznego, który ma określone właściwości fizyczne i chemiczne i jest oddzielony od innych części układu interfejsem.
W prawdziwych roztworach substancja jest rozdrabniana do stanu molekularnego i nie ma granicy między substancją rozpuszczoną a rozpuszczalnikiem.
Przyczyna zjawisk powierzchniowych to istnienie na styku stykających się faz nienasyconego pola sił międzyatomowych, międzycząsteczkowych, które powstaje w wyniku inny skład oraz strukturę sąsiednich faz i różnice w wiązaniach ich atomów i cząsteczek na powierzchni.
Warstwy powierzchniowe ciał ciekłych i stałych przylegające do granicy faz różnią się znacznie pod względem wielu wskaźników fizycznych i chemicznych od właściwości faz w głębi ich objętości (energia właściwa, gęstość, lepkość, właściwa przewodność elektryczna itp.). Różnice związane są także z pewną orientacją cząsteczek w warstwach powierzchniowych i ich odmiennym stanem energetycznym w porównaniu z cząsteczkami w masie. Ponadto w układach wieloskładnikowych (roztworach) skład warstwy powierzchniowej nie pokrywa się ze składem faz objętościowych.
Cechy warstw powierzchniowych wynikają z obecności nadmiaru energii powierzchniowej. Właściwości interfejsu mają tym większy wpływ na zachowanie systemu jako całości większy obszar powierzchnie (uderzenie S). Wyjaśnia to dominującą rolę zjawisk powierzchniowych we właściwościach układów silnie rozproszonych, których Ssp osiąga ogromne wartości.
Obecność nadmiaru energii w warstwie powierzchniowej cząsteczek wynika z niepełnej kompensacji międzycząsteczkowych sił przyciągania pomiędzy cząsteczkami warstwy powierzchniowej ze względu na ich słaba interakcja z sąsiednią fazą.
Badania chemii koloidalnej systemy rozproszone – układy heterogeniczne składające się z dwóch lub więcej faz, z których jedna faza rozproszona - fragmentaryczne (nieciągłe), a drugie - ośrodek dyspersyjny - jest ciągłą częścią systemu.
Pojęcie mikroheterogenicznego charakteru roztworów koloidalnych i innych układów rozproszonych ma fundamentalne znaczenie. Za swoje odkrycie austriacki naukowiec Zsigmondy (*) został laureatem nagroda Nobla chemii w 1925 r
Wybór w specjalna grupa rozproszone cząstki są spowodowane różnicą w ich właściwościach fizycznych i właściwości chemiczne z podobnych właściwości dużych obiektów tej samej substancji. Właściwości te obejmują wytrzymałość, pojemność cieplną, T pl, magnetyczne i Parametry elektryczne, reaktywność.
Różnice te wynikają z efektów wielkości. Im mniejszy rozmiar cząstek, tym wyraźniejsze są specjalne właściwości, szczególnie dotyczy to nanocząstek. Właściwości te otwierają zasadniczo nowe zastosowania praktyczne w chemii, fizyce i biologii. Badanie właściwości cząstek rozproszonych (metody wytwarzania, struktury, fizyki i chemii) jest jednym z najpilniejszych i najbardziej obiecujących zadań w wielu dyscyplinach.
Zdyspergowane cząstki mogą mieć bardzo różne właściwości formularz : cylindryczne, kuliste, prostokątne, nieregularne. Na przykład rozproszone cząstki obejmują:
układy z cząstkami sześciennymi, kulistymi - zole, emulsje, zawiesiny, pasty;
nitkowate – włókna komórek nerwowych, dwuwymiarowe włókna mięśniowe, naczynia włosowate, pory (drewno, tkanki, włosy, skóra),
folie - warstwy powierzchniowe na granicy faz w emulsjach, piankach, w porach katalizatorów i adsorbentów, membrany.
Zatem 1 m 3 materiał wyjściowy można pokroić w kostkę o długości krawędzi A, przeciągnij w nić o przekroju A lub spłaszczyć w grubą warstwę A.
Jeśli cząstki mają nieregularny kształt, to posługując się pojęciem „wielkości poprzecznej”, ich kształt utożsamia się z kulistym o równoważnej średnicy.
Charakterystyka ilościowa układu dyspersyjnego:
1. Wielkość cząstek d śr., d min., d max
2. Stężenie cząstek ν = n d /V, gdzie n d jest liczbą cząstek fazy rozproszonej na jednostkę objętości ośrodka dyspersyjnego V
3. Fragmentacja systemu charakteryzuje się rozproszeniem D I powierzchnia właściwa fazy rozproszonej Ssp:
Pierwszą opcją oceny ilościowej jest podstawowy
D= 1/d I S uderzenie = S / V,(1.1)
Gdzie D – minimalny rozmiar cząstki, S - całkowita powierzchnia międzyfazowa, V- objętość ciała.
Współczesna chemia koloidalna jest nauką z pogranicza chemii, fizyki i biologii. Szczególną interdyscyplinarną pozycję chemii koloidów podkreśla fakt, że w literaturze anglojęzycznej często używa się nazwy „nauka o koloidach”. nauka o koloidach).
Historia chemii koloidalnej
Chemia koloidów jako nauka mała historia Jednak już od czasów starożytnych ludzie wykorzystywali właściwości układów koloidalnych i procesów koloidowo-chemicznych. Są to na przykład takie rzemiosła, jak wyrób farb, ceramiki, glazury, przędzenie lnu, bawełny, wełny, garbowanie skór.
Począwszy od XVIII wieku pojawiały się opisy poszczególnych badań, które później włączano do odpowiednich działów chemii koloidów. Należą do nich prace M.V. Łomonosowa dotyczące krystalizacji i produkcji kolorowych szkieł przy użyciu dyspersji metali (1745-1755). Niezależnie od siebie K. Scheele i F. Fontana odkryli zjawisko adsorpcji gazu przez węgiel. W T. E. Lovitz odkrył zjawisko adsorpcji z roztworów. Pierwszą otrzymał P. Laplace relacje ilościowe dla ciśnienia kapilarnego. W 1808 roku F. F. Reiss, przeprowadzając eksperymenty z pierwiastkiem Volty, odkrył zjawiska elektroforezy i elektroosmozy.
Jedne z najwcześniejszych badań układów koloidalnych przeprowadził Włoch F. Selmi w 1845 roku. Badał układy składające się z chlorku srebra, siarki i błękitu pruskiego rozmieszczonych w objętości wody. Układy te otrzymane przez Selmiego są bardzo podobne do roztworów prawdziwych, jednak Selmi uważał, że ani badane przez niego substancje, ani inne podobne substancje nie mogą występować w wodzie w postaci tych samych małych cząstek, jakie powstają w roztworach prawdziwych, czyli w w postaci pojedynczych cząsteczek lub jonów.
Poglądy bliskie Selmiemu wyrażał K. Nägeli, który uważał, że w takich układach cząstki siarki, chlorku srebra i innych substancji stanowią większe agregaty niż pojedyncze cząsteczki. Dla agregatów wielocząsteczkowych wprowadził pojęcie „miceli”. Aby odróżnić układy zawierające micele od roztworów, w których substancja rozpuszczona występuje w postaci pojedynczych cząsteczek, Nägeli nazwał układy zawierające micele „solami”. Terminy „micela” i „sol” stały się powszechnie akceptowane.
Stan aktulany
Główne kierunki współczesnej chemii koloidów:
- Termodynamika zjawisk powierzchniowych.
- Badanie adsorpcji surfaktantów.
- Badanie powstawania i stabilności układów dyspersyjnych, ich właściwości molekularno-kinetycznych, optycznych i elektrycznych.
- Mechanika fizykochemiczna struktur rozproszonych.
- Opracowanie teorii i mechanizmów molekularnych procesów zachodzących w układach rozproszonych pod wpływem surfaktantów, ładunki elektryczne, uderzenia mechaniczne itp.
Ponieważ rozproszony stan materii jest uniwersalny, a przedmioty badań chemii koloidalnej są bardzo różnorodne, chemia koloidalna jest ściśle powiązana z fizyką, biologią, geologią, gleboznawstwem, medycyną itp.
Jego imieniem nazwano Instytut Chemii Koloidów i Chemii Wody. A. V. Dumansky NASU (Kijów).
Ukazuje się naukowe czasopismo „Colloid Journal”.
Literatura
- Podręcznik chemii powierzchni i koloidów / wyd. K.S. Ptaszek. - wyd. 2 - NY: CRC Press, 2003. - 765 s.
- Ablesimov N. E. Streszczenie chemii: Podręcznik i podręcznik chemii ogólnej - Chabarowsk: Wydawnictwo FEGUPS, 2005. - 84 s.
- Ablesimov N. E. Ile chemii jest na świecie? Część 1. // Chemia i życie - XXI wiek. - 2009. - nr 5. - s. 49-52.
- Summ B. D. Podstawy chemii koloidalnej: podręcznik. pomoc dla studentów wyższy podręcznik instytucje / B.D. Sum. - wyd. 2, skreślone. - M.: Centrum wydawnicze„Akademia”, 2007. - 240 s.
- Encyklopedia chemiczna. - M.: "BRE", 1998.
- Friedrichsberg D. A. Kurs chemii koloidów. L.: Chemia, 1984. - 352 s.
- Zakharchenko V.N. Chemia koloidalna: podręcznik. dla biologa medycznego. specjalista. uniwersytety - wyd. 2, poprawione. i dodatkowe - M.: Szkoła wyższa, 1989.-238 s.: il.
Fundacja Wikimedia. 2010.
Zobacz, co oznacza „chemia koloidalna” w innych słownikach:
CHEMIA KOLOIDALNA, bada układy rozproszone, które mają wysoki stopień fragmentacja (wielkość cząstek od 10 2 do 10 7 cm) i ogromna powierzchnia (przykładowo węgiel aktywny ma powierzchnię właściwą tysięcy m2/g), która je determinuje... ... Nowoczesna encyklopedia
chemia koloidów- - dział chemii, którego przedmiotem są układy silnie rozproszone i układy w nich przepływające. Słownik chemii analitycznej... Terminy chemiczne
CHEMIA KOLOIDÓW- nauka zajmująca się fizyką. chemia właściwości układów rozproszonych i niektórych produktów wielkocząsteczkowych, a także powierzchniowe zjawiska fizyczne. chemia procesy zachodzące na interfejsie (patrz) ... Wielka encyklopedia politechniczna
Tradycyjna nazwa chemii fizycznej układów rozproszonych (patrz Układy rozproszone) i zjawisk powierzchniowych (patrz Zjawiska powierzchniowe). K. x. jako samodzielna nauka powstała w latach 60. XIX wieku. Od tego czasu jego przedmiot i metody znacznie się zmieniły... Wielka encyklopedia radziecka
Termin chemia koloidów Termin w języku angielskim chemia koloidów Synonimy nauka o koloidach Skróty Powiązane terminy adhezja, adsorpcja, podwójna warstwa elektryczna, dyspersja, zol, roztwór koloidalny, stężenie krytyczne... ... Encyklopedyczny słownik nanotechnologii
Dziedzina chemii zajmująca się badaniem układów rozproszonych i zjawisk powierzchniowych zachodzących na granicach faz. Ponieważ cząstki fazy rozproszonej i otaczający je ośrodek dyspersyjny mają bardzo dużą powierzchnię międzyfazową (w układach silnie rozproszonych... ... Encyklopedia chemiczna
Tradycyjna nazwa nauki o układach rozproszonych i zjawiskach powierzchniowych. Bada procesy i zjawiska takie jak adhezja, adsorpcja, zwilżanie, koagulacja, elektroforeza. Opracowuje naukowe podstawy technologii materiałów budowlanych, wiercenia... słownik encyklopedyczny
chemia koloidów- koloidų chemija statusas T sritis chemija apibrėžtis Dispersinių sistemų ir paviršinių reiškinių chemija. atitikmenys: pol. chemia koloidów rus. chemia koloidalna... Chemijos terminų aiškinamasis žodynas
Nauka o zjawiskach powierzchniowych i układach rozproszonych. Cała natura skorupa Ziemska i podłoże, atmosfera i hydrosfera, organizmy zwierzęce i roślinne, złożony zestaw różnorodnych systemów rozproszonych. Powszechność stanu rozproszonego determinuje... ... Wielki encyklopedyczny słownik politechniczny
Książki
- Chemia koloidalna. Fizykochemia układów dyspersyjnych. Podręcznik dla studentów wyższych uczelni zawodowych. Grif Ministerstwo Obrony Federacji Rosyjskiej Erszow Jurij Aleksiejewicz. W podręczniku przedstawiono podstawy chemii fizycznej układów rozproszonych (chemia koloidalna) zgodnie z przybliżony program w dyscyplinie „Chemia fizyczna i koloidalna” dla specjalności 060301...