Ecocardiografia na drenagem anômala das veias pulmonares. Drenagem venosa pulmonar anômala parcial Drenagem venosa pulmonar anormal Diagnóstico
Leia também
10 U d/m 2 inicialmente e > 7 U d/m 2 após o uso de vasodilatadores); - a presença de contra-indicações absolutas para patologia somática concomitante. Táticas cirúrgicas Na ausência de sintomas, a correção deve ser realizada antes dos 3-4 anos de idade. A correção oportuna ajuda a evitar complicações a longo prazo da VPPB: hipertensão pulmonar e insuficiência ventricular direita, flutter e fibrilação atrial. Pacientes com síndrome da Cimitarra desenvolvem sintomas de insuficiência cardíaca durante a infância. O tratamento cirúrgico é estadiado - primeiramente, é realizado o cateterismo cardíaco para detectar e embolizar as colaterais aortopulmonares para o pulmão direito. Além disso, dependendo dos sintomas de insuficiência cardíaca, é realizada a correção cirúrgica da PALV. Técnica cirúrgica Se a CIA for pequena ou ausente, ela é expandida ou moldada. O deslocamento da boca do PV na cavidade do RA é realizado por um remendo, é costurado para não estreitar a boca do PV. Em caso de estenose da veia cava, uma plastia adicional de sua boca é realizada com um adesivo autoxenopericárdico. Técnicas para correção de VPVP: - isolamento e transplante de VPs com fluxo anormal para o átrio esquerdo ou para VPs com fluxo normal; - divisão do tronco comum da veia cava superior ou inferior em dois canais (coletor de veia pulmonar e coletor de veia cava) com posterior direcionamento de seus fluxos para os átrios; - anastomose da extremidade distal da VCS (a VP é drenada pelo segmento proximal da VCS); - comutação intracardíaca por ASD de correntes venosas dos grandes e pequenos círculos de circulação sanguínea; Como material para plásticos, uso tautopericárdio tratado com glutaraldeído, ou xenopericárdio. Complicações específicas do tratamento cirúrgico: - shunt residual entre os átrios; - estenoses das bocas das veias ocas ou veias pulmonares; - síndrome de fraqueza do nó sinusal na plástica da boca do VIV; NRS, taquiarritmias. Observação pós-operatória 1. A duração da observação de pacientes com AFVD corrigida na ausência de distúrbios hemodinâmicos não é superior a 2 anos. Antes do cancelamento do registro, são realizados um ECG e um exame de ultrassom do coração. 2. Em caso de registro no pós-operatório de NRS (bradiarritmias, taquicardias atriais), além do exame, recomenda-se o SMEKG a cada 6 meses ou mais. Se indicado, é realizada terapia antiarrítmica, RFA ou implante de marcapasso. 3. A prevenção da endocardite bacteriana é realizada de acordo com as indicações nos primeiros 6 meses após a correção cirúrgica do defeito. 4. A admissibilidade de educação física e esportes após a correção do defeito. (max-width: 448px) 100vw, 448px"> PALV é caracterizada por uma ou mais, mas não todas, veias pulmonares drenando para o átrio direito e suas tributárias As veias pulmonares podem drenar para a veia inominada, seio coronário, VCS (veia cava superior), PP (átrio direito), VCI (veia cava inferior), veias porta, veias hepáticas.
Ao contrário da TADPV (drenagem venosa pulmonar anômala total), esse defeito pode ocorrer sem combinação com CIA (defeito do septo atrial) (5,7%).
As variantes clinicamente mais significativas de PALV são:
- veias dos lobos superiores e/ou médios do pulmão direito desembocam na VCS abaixo da boca da veia inominada (combinadas com CIA "sinus venosus");
- as veias do lobo inferior do pulmão direito fluem para a VCI acima ou abaixo do nível do diafragma (faz parte da "síndrome da cimitarra"
- suprimento sanguíneo arterial anormal e hipoplasia do lobo inferior do pulmão direito, dextroposição do coração);
- a confluência da VP esquerda (veia(s) pulmonar(es)) na veia inominada através da veia vertical esquerda.
A hemodinâmica da PALV é em muitos aspectos semelhante à CIA, com a única diferença de que a hipervolemia da CCI (circulação pulmonar) está associada não ao shunt esquerdo-direito, mas à sua recirculação parcial pelos pulmões. O volume de recirculação depende do número de veias pulmonares drenantes, da presença de DMMP e da magnitude do TRL (resistência pulmonar total). A taxa de desenvolvimento de HP (hipertensão pulmonar) depende do volume de sangue que recircula pelos pulmões. A HP atinge um grau significativo na 3ª-4ª década de vida.
CONSULTÓRIO
uma. Manifestações clínicas da doença:
- as crianças com PADL são geralmente assintomáticas;
- na presença de drenagem da VP direita na VCI, as crianças podem apresentar infecções broncopulmonares frequentes.
b. Exame físico: divisão permanente do II tom no II m.r. à esquerda do esterno, independente das fases da respiração (com combinação de CHADLV e CIA);
sopro sistólico de intensidade fraca ou moderada (não superior a 3/6) de estenose valvar relativa do AE em II m. à esquerda do esterno;
Sopro mesodiastólico suave de estenose relativa da CT (valva tricúspide);
Ao longo da borda esquerda do esterno no terço inferior.
DIAGNÓSTICO
- Eletrocardiografia
- hipertrofia do pâncreas (ventrículo direito);
- bloqueio completo ou incompleto da perna direita do feixe de His;
- a variante normal do ECG não é excluída.
- ecocardiografia
O diagnóstico de PALV geralmente é obtido pela detecção da ausência da confluência típica de uma ou mais veias pulmonares no AE em locais típicos.
ASDs altos do tipo "sinus venosus", como regra, são combinados com PALV.
Critério de diagnóstico:
- dilatação dos departamentos certos;
- expansão da artéria pulmonar;
- diminuição relativa em LA e LV;
- veia cava superior dilatada.
TRATAMENTO E OBSERVAÇÃO
- Observação e tratamento de pacientes com PADL não corrigida
uma. A prevenção da endocardite bacteriana não está indicada.
b. Correção de sintomas de insuficiência cardíaca (em casos raros).
dentro. A admissibilidade da educação física no esporte antes da correção do defeito.
- Cirurgia
Indicações para tratamento cirúrgico
O diagnóstico confirmado de PADLV é indicação absoluta para tratamento cirúrgico.
Contra-indicações para o tratamento cirúrgico:
- hipertensão pulmonar elevada (ARS > 10 U d/m 2 inicialmente e > 7 U d/m 2 após o uso de vasodilatadores);
- a presença de contra-indicações absolutas para patologia somática concomitante.
Táticas cirúrgicas
Na ausência de sintomas, a correção deve ser realizada antes dos 3-4 anos de idade. A correção oportuna ajuda a evitar complicações a longo prazo da VPPB: hipertensão pulmonar e insuficiência ventricular direita, flutter e fibrilação atrial.
Em pacientes com síndrome da cimitarra sintomas de insuficiência cardíaca desenvolvem-se na infância. O tratamento cirúrgico é estadiado - primeiramente, é realizado o cateterismo cardíaco para detectar e embolizar as colaterais aortopulmonares para o pulmão direito. Além disso, dependendo dos sintomas de insuficiência cardíaca, é realizada a correção cirúrgica da PALV.
Técnica cirúrgica
No caso de tamanho pequeno ou ausência de ASD, ele é expandido ou formado. O deslocamento da boca do PV na cavidade do RA é realizado por um remendo, é costurado para não estreitar a boca do PV. Em caso de estenose da veia cava, uma plastia adicional de sua boca é realizada com um adesivo autoxenopericárdico.
Técnicas para corrigir PHADLV:
- isolamento e transplante de VPs com fluxo anormal para o átrio esquerdo ou para VPs com fluxo normal;
- divisão do tronco comum da veia cava superior ou inferior em dois canais (coletor de veia pulmonar e coletor de veia cava) com posterior direcionamento de seus fluxos para os átrios;
- anastomose da extremidade distal da VCS (a VP é drenada pelo segmento proximal da VCS);
- comutação intracardíaca por ASD de correntes venosas dos grandes e pequenos círculos de circulação sanguínea;
Como material para plásticos, uso tautopericárdio tratado com glutaraldeído, ou xenopericárdio.
Complicações específicas do tratamento cirúrgico:
- derivação residual entre os átrios;
- estenose das bocas das veias ocas ou veias pulmonares;
- síndrome de fraqueza do nó sinusal com cirurgia plástica do orifício do VIV; NRS, taquiarritmias.
Acompanhamento pós-operatório
- A duração da observação de pacientes com PADL corrigida na ausência de distúrbios hemodinâmicos não é superior a 2 anos. Antes do cancelamento do registro, é realizado um exame de ultrassom do coração.
- Em caso de registro no pós-operatório de NRS (bradiarritmias, taquicardias atriais), além do exame, recomenda-se SMEKG a cada 6 meses ou mais frequentemente. Se indicado, é realizada terapia antiarrítmica, RFA ou implante de marcapasso.
- A prevenção da endocardite bacteriana é realizada de acordo com as indicações nos primeiros 6 meses após a correção cirúrgica do defeito.
- A admissibilidade de educação física e esportes após a correção do defeito.
A veia pulmonar (foto abaixo) é um vaso que leva sangue arterial, enriquecido com oxigênio nos pulmões, para o átrio esquerdo.
Partindo dos capilares pulmonares, esses vasos se fundem em veias maiores, que vão para os brônquios, depois segmentos, lobos e nas portas do pulmão formam grandes troncos (dois de cada raiz), que em posição horizontal vão para a parte superior parte do átrio esquerdo. Nesse caso, cada um dos troncos penetra em um orifício separado: os esquerdos - do lado esquerdo do átrio esquerdo e os direitos do direito. As veias pulmonares direitas, seguindo para o átrio (esquerdo), cruzam transversalmente o átrio direito (sua parede posterior).
Veia pulmonar superior (direita)
É formado por veias segmentares de segmentos dos lobos médio e superior do pulmão.

Veia pulmonar inferior (direita)
Este vaso recebe sangue do lobo inferior (seus 5 segmentos) e possui dois influxos principais: a veia basal comum e o ramo superior.
Ramo superior
Situa-se entre os segmentos basal e superior. É formado a partir das veias acessórias e principais, segue para frente e para baixo, passando por trás do brônquio apical segmentar. Este ramo é o mais alto de todos os que desembocam na veia pulmonar inferior direita.
Correspondendo ao brônquio, a veia principal contém três tributárias: lateral, superior, medial, localizada principalmente intersegmentarmente, mas também pode situar-se intrasegmentarmente.

Graças à veia acessória, o sangue é drenado do segmento superior (sua parte superior) para a região sublobar da veia segmentar posterior do lobo superior (seu segmento posterior).
Veia comum basal
É um tronco venoso curto formado pela confluência das veias basais inferior e superior, cujos ramos principais se situam muito mais profundamente que a superfície lobar anterior.
Veia basal superior. É formado devido à confluência das maiores veias segmentares basais, bem como veias que transportam sangue dos segmentos medial, anterior e lateral.
Veia basal inferior. Adjacente à veia comum basal do lado de sua superfície posterior. O principal tributário desse vaso é o ramo posterior basal, que coleta sangue do segmento basal posterior. Em alguns casos, a veia basal inferior pode se aproximar da veia basal superior.
ADLV
É uma patologia congênita do coração, na qual se detecta uma entrada não anatômica das veias pulmonares no átrio (direita) ou nas últimas veias ocas.

Esta patologia é acompanhada por pneumonia frequente, fadiga, falta de ar, atraso no desenvolvimento físico, dor no coração. Como diagnóstico, eles usam: ECG, ressonância magnética, radiografia, ultra-som, ventriculo- e atriografia, angiopulmonografia.
O tratamento cirúrgico do defeito depende do seu tipo.
Informação geral
O ADLV é um defeito congênito e representa cerca de 1,5-3,0% dos defeitos cardíacos. Observado principalmente em pacientes do sexo masculino.
Na maioria das vezes, esse defeito é combinado com uma janela oval (aberta) e defeitos septais entre os ventrículos. Um pouco menos frequentemente (20%) - com um tronco comum arterial, hipoplasia do lado esquerdo do coração, CIV, dextrocardia, tétrade de Fallot e transposições dos vasos principais, um ventrículo comum do coração.
Além dos defeitos acima, o ADLV é frequentemente acompanhado por patologia extracardíaca: hérnias umbilicais, malformações dos sistemas endócrino e ósseo, divertículos intestinais, rim em ferradura, hidronefrose e doença renal policística.
Classificação da drenagem venosa pulmonar anômala (APLV)
Se todas as veias fluem para a circulação sistêmica ou para o átrio direito, esse defeito é chamado de drenagem anômala completa, mas se uma ou mais veias fluem para as estruturas acima, esse defeito é chamado de parcial.
De acordo com o nível de confluência, várias variantes do defeito são distinguidas:
- Opção um: supracardial (supracardial). As veias pulmonares (como um tronco comum ou separadamente) desembocam em seus ramos.
- Opção dois: cardíaca (intracardíaca). As veias pulmonares são drenadas para o átrio direito.
- Opção três: subcardíaco (infra ou subcardíaco). As veias pulmonares entram na veia cava porta ou inferior (com muito menos frequência no ducto linfático).
- Quarta opção: misto. As veias pulmonares entram em várias estruturas e em diferentes níveis.
Características da hemodinâmica
No período intrauterino, esse defeito, via de regra, não se manifesta, devido às peculiaridades da circulação sanguínea do feto. Após o nascimento de um bebê, as manifestações de distúrbios hemodinâmicos são determinadas pela variante do defeito e sua combinação com outras anomalias congênitas.
No caso de drenagem anômala total, os distúrbios hemodinâmicos são expressos por hipoxemia, sobrecarga hipercinética do coração direito e hipertensão pulmonar.
No caso de drenagem parcial, a hemodinâmica é semelhante à do CIA. O papel principal nas violações pertence à descarga venoso-arterial anormal de sangue, o que leva a um aumento do volume sanguíneo no pequeno círculo.
Sintomas de drenagem venosa pulmonar anormal
As crianças com este defeito muitas vezes sofrem de SARS repetida e pneumonia, têm tosse, baixo ganho de peso, taquicardia, falta de ar, dor no coração, cianose leve e fadiga.

No caso de hipertensão pulmonar óbvia em idade jovem, insuficiência cardíaca, cianose grave e
Diagnóstico
O quadro de auscultação em ADLV é semelhante ao da CIA, ou seja, ouve-se um sopro sistólico rugoso na área de projeções das artérias das veias (veias pulmonares) e desdobramento do 2º tom.
- O ECG mostra sinais de sobrecarga do coração direito, desvio EOS para a direita, bloqueio (incompleto) da perna direita do feixe de Hiss.
- A fonografia mostra sinais de TEA.
- Na radiografia, o padrão dos pulmões é realçado, a artéria pulmonar (seu arco) se projeta, a expansão das bordas cardíacas para a direita, o sintoma "sabre turco".
- EcoCG.
- Sondagem das cavidades cardíacas.
- Flebografia.
- Atriografia (direita).
- Angiopneumografia.
- Ventriculografia.

O diagnóstico diferencial deste defeito deve ser realizado com:
- Linfangiectasia.
- Atresia
- Transposição vascular.
- Estenose mitral.
- Estenose das veias pulmonares direita/esquerda.
- Coração triplo.
- TEA isolado.
Tratamento
Os tipos de tratamento cirúrgico da drenagem parcial são determinados pela variante do defeito, tamanho e localização da CIA.

A comunicação interatrial é eliminada com a ajuda de plásticos ou sutura da CIA. Bebês de até três meses de idade, que estão em estado crítico, são submetidos a uma operação paliativa (atriosepotomia fechada), que visa ampliar a comunicação interatrial.
A correção radical geral do defeito (forma total) inclui várias manipulações.
- Ligadura da comunicação patológica de vasos com veias.
- Isolamento das veias pulmonares.
- Fechamento do DMP.
- Formação de uma anastomose entre o átrio esquerdo e as veias pulmonares.
A consequência de tais operações pode ser: aumento da hipertensão pulmonar e síndrome da insuficiência do nó sinusal.
Previsões
O prognóstico para o curso natural desse defeito é desfavorável, pois 80% dos pacientes morrem durante o primeiro ano de vida.
Pacientes com drenagem parcial podem viver até os trinta anos. A morte desses pacientes é mais frequentemente associada a infecções pulmonares ou insuficiência cardíaca grave.
Os resultados das correções cirúrgicas do defeito costumam ser satisfatórios, mas entre os recém-nascidos, a mortalidade durante ou após a cirurgia permanece alta.
As veias pulmonares desembocam no átrio esquerdo, e é através delas que o sangue oxigenado entra no ventrículo esquerdo e depois na aorta. Além disso, é transportado por artérias por toda a circulação sistêmica.
Sob a influência de vários fatores negativos externos no período pré-natal, podem ser criadas condições para a formação de um defeito cardíaco congênito como drenagem venosa pulmonar anormal (ADPV), na qual esses vasos fluem não para a esquerda, mas para o átrio direito, veia cava ou seio coronário. Essa localização anormal dos vasos pode ser parcial ou total, ou seja, parte das veias pulmonares individuais ou todas as bocas desses vasos podem se comunicar com o átrio direito. A condição do paciente depende em grande parte desses fatores.
Segundo as estatísticas, o ADLV é detectado em 1,5-3% dos pacientes com. Mais frequentemente esta patologia encontra-se entre os homens.
Muitas vezes, o ADLV é combinado com tais anomalias no desenvolvimento do coração como um defeito do septo atrial. E em 20% dos pacientes, é acompanhado por outros defeitos: tétrade de Fallot, defeito do septo ventricular, subdesenvolvimento das câmaras esquerdas do coração, etc. Além disso, anomalias de desenvolvimento não cardíacas são frequentemente detectadas em pacientes com este diagnóstico: ferradura rim em forma, doença renal policística, hidronefrose, hérnia umbilical, divertículos intestinais e malformações do sistema musculoesquelético ou do sistema endócrino.
Neste artigo, apresentaremos as possíveis causas, tipos, manifestações, métodos para detectar e tratar a drenagem venosa pulmonar anormal. As informações obtidas ajudarão você a entender a essência dessa cardiopatia congênita e poderá fazer ao seu médico perguntas que lhe interessem.
Essa rara malformação congênita foi descrita pela primeira vez por Wilson em 1798, mas suas variantes foram consideradas com mais detalhes somente após o acúmulo de experiência cirúrgica cardíaca suficiente nas décadas de 1950 e 1960. A primeira operação bem sucedida para corrigir esta patologia foi realizada por Miller em 1951. Posteriormente, as técnicas cirúrgicas cardíacas foram aprimoradas e, em 1956, foi realizada uma correção radical da ADLV com hipotermia superficial. No mesmo ano, uma intervenção semelhante foi realizada com circulação extracorpórea.
No território da URSS, pela primeira vez, uma operação bem-sucedida para eliminar esse defeito cardíaco foi realizada por Nikolai Mikhailovich Amosov em 1978, e um dos principais cirurgiões cardíacos russos, Georgy Eduardovich Falkovsky, relatou uma série de correções bem-sucedidas em crianças pequenas em 1984.
As razões
Provavelmente, a hereditariedade desempenha um certo papel no desenvolvimento de ADLV.As prováveis causas do desenvolvimento de ADLV podem ser os mesmos fatores externos que provocam outras anomalias congênitas na estrutura do coração e vasos sanguíneos:
- hereditariedade (mutações de cromossomos);
- tomar certos medicamentos teratogênicos;
- exposição a substâncias tóxicas no corpo durante a gravidez;
- maus hábitos da futura mãe;
- doenças infecciosas transmitidas por uma mulher grávida;
- distúrbios endócrinos;
- toxicose;
- ambiente desfavorável.
A dissociação das veias pulmonares com o átrio esquerdo pode ser causada pelos seguintes fatores:
- a ausência de sua conexão com o átrio esquerdo - é formada devido ao fato de que, sob a influência das causas externas descritas acima, o crescimento atrial esquerdo não pode entrar em contato com os plexos venosos do futuro pulmão;
- atresia precoce da veia pulmonar - ocorre com a conexão inicial do leito vascular pulmonar e da veia pulmonar comum, cujo lúmen é posteriormente obliterado, e o sangue dos pulmões começa a fluir por outras vias colaterais.
Variedades
ADLV é de dois tipos:
- Total (ou completo). Com esse defeito, a circulação sanguínea entre os grandes e pequenos círculos para completamente, pois não há comunicação entre as veias pulmonares e o átrio esquerdo. Essa forma de defeito é incompatível com a vida, no entanto, a criança pode ter anomalias concomitantes no desenvolvimento do coração como defeitos no septo interatrial ou interventricular, compensando levemente uma violação tão significativa da hemodinâmica. Nesses casos, o bebê pode viver por algum tempo. No entanto, na maioria dos casos, sem correção cirúrgica cardíaca oportuna do ADLV total, as crianças morrem antes do primeiro ano de vida.
- Parcial. Com tal anomalia, um dos ramos das veias pulmonares se comunica com o átrio esquerdo, e o estado hemodinâmico do paciente depende do volume de descarga sanguínea.
Dependendo do nível de entrada das veias pulmonares no átrio esquerdo, distinguem-se quatro variantes anatômicas do ADLV:
- supracárdica (ou supracárdica) - observada em quase 55% dos recém-nascidos e acompanhada pelo fluxo das veias pulmonares para o sistema da veia cava superior;
- cardíaca (ou intracardíaca) - detectada em 30% dos pacientes e acompanhada pelo fluxo das veias pulmonares para o átrio direito ou seio coronário;
- subcardíaco (ou subcardíaco, infracardíaco) - observado em 12% das crianças e é acompanhado pelo fluxo de veias pulmonares para a veia cava inferior ou veia porta (às vezes para o ducto linfático);
- misto - detectado em 3% dos recém-nascidos e pode ser acompanhado por diversas variações da confluência das veias pulmonares descritas acima.
Como a hemodinâmica é perturbada?
Durante o desenvolvimento intrauterino, essa cardiopatia não se manifesta de forma alguma, pois a circulação intracardíaca no feto é acompanhada pela comunicação dos átrios esquerdo e direito pela janela oval aberta. O ADLV começa a se fazer sentir após o nascimento de uma criança, e a gravidade dos distúrbios hemodinâmicos será determinada pelo tipo e variante dessa anomalia. Além disso, a presença de defeitos concomitantes no desenvolvimento do coração e dos vasos sanguíneos pode afetar a gravidade dos sintomas do defeito.
Com o ADLV total, o sangue enriquecido com oxigênio que entra no átrio direito se mistura com o sangue venoso. Além disso, parte dele entra no ventrículo direito e o outro entra no átrio esquerdo através de um defeito do septo atrial existente ou de um forame oval aberto. Nesses casos, o ADLV total é compatível com a vida, pois não se perde a comunicação entre a circulação sistêmica e pulmonar. Tais distúrbios hemodinâmicos levam a uma sobrecarga das câmaras direitas do coração, aumento da pressão nos vasos pulmonares e diminuição do conteúdo de oxigênio no sangue, o que leva à falta de oxigênio de órgãos e tecidos.
Com ADLV parcial, os distúrbios hemodinâmicos ocorrem da mesma forma que nos defeitos interatriais. A gravidade da condição do paciente com esse tipo de anomalia é determinada pela quantidade de descarga arteriovenosa patológica.
Sintomas
Sinais de ADLV se fazem sentir imediatamente após o nascimento de uma criança. Com um defeito total, que não é acompanhado pela presença de defeitos nas comunicações interatriais, a circulação sanguínea entre o pequeno e o grande círculo torna-se completamente impossível e o recém-nascido morre rapidamente. Nesses casos, a vida da criança só pode ser salva por uma cirurgia cardíaca de emergência usando o método de Rashkind como septostomia atrial endovascular por balão.
Em outros casos, a gravidade das manifestações clínicas de ADLV é determinada por sua variante anatômica, o tamanho dos defeitos do septo atrial e a natureza dos distúrbios hemodinâmicos. Normalmente, os pais de crianças com esse defeito congênito notam as seguintes manifestações dessa patologia:
- fatigabilidade rápida;
- o aparecimento de falta de ar após atividade física (alimentação, choro, etc.);
- dor na região do coração (a criança não dorme bem, fica inquieta, chora alto e estica as pernas);
- pele pálida;
- leve ;
- tosse;
- nausea e vomito;
- ganho de peso lento
- SARS e pneumonia frequentes.
Ao sondar o pulso, o médico pode detectar sua rapidez e arritmia. No exame, deformidade torácica e aumento do batimento cardíaco são determinados.
A cianose leve que ocorre nas primeiras semanas de vida pode desaparecer ou tornar-se menos pronunciada ao longo do tempo. Como regra, a cianose é mais pronunciada durante o esforço físico.
Ao ouvir os sons do coração, os sinais específicos desse defeito não são detectados. Pode ser ouvido:
- alto tom sobre o coração;
- desdobramento do tom II com aumento do componente pulmonar;
- tom III auscultado sobre o ápice do coração (em muitos pacientes);
- sopro sistólico suave sobre a artéria pulmonar com duração e intensidade variáveis.
No ECG, uma sobrecarga do coração direito é determinada - uma alta voltagem da onda P nas derivações direitas e desvios do eixo elétrico para a direita nas derivações padrão. Uma sobrecarga do átrio direito é indicada por uma onda P alta no tórax direito e derivações padrão. Muitas vezes há sinais de incompleta.
Se houver um grande defeito no septo interatrial, a criança pode se desenvolver normalmente e, em uma idade mais avançada, há queixas de uma diminuição significativa da tolerância ao exercício, falta de ar, palidez e problemas de saúde geral. Mais tarde, essas crianças desenvolvem sintomas.
Diagnóstico
 Um dos métodos para ajudar a detectar e verificar o ADLV é o ultrassom cardíaco.
Um dos métodos para ajudar a detectar e verificar o ADLV é o ultrassom cardíaco. Pela primeira vez, o ADLV pode ser detectado no feto durante um exame de ultra-som no terceiro trimestre de gravidez.
Para esclarecer todos os dados sobre essa doença cardíaca congênita, são prescritos os seguintes métodos instrumentais de pesquisa:
- radiografia;
- (adultos e crianças maiores são prescritos);
- sondagem das câmaras do coração e.
Tratamento
A única forma de eliminar o ADLV é sua correção cardiocirúrgica. Antes da cirurgia, recomenda-se que a criança limite a atividade física e prescrevem-se medicamentos para prevenir a insuficiência cardíaca aguda (,). Os pais de uma criança devem estar cientes de que mesmo o choro ou o desconforto da temperatura podem piorar significativamente o bem-estar com essa doença cardíaca. Se a criança já é adulta, eles devem monitorar constantemente seu tempo de lazer - ele não deve correr, levantar pesos e trabalhar demais.
Métodos de correção cirúrgica
O método de realização da correção cirúrgica cardíaca em ADLV é determinado pelo tipo e variante do defeito.
Com a drenagem total, uma criança criticamente enferma com menos de 3 meses de idade pode ser submetida a uma operação paliativa, que consiste em aumentar a comunicação interatrial através da realização de atriosseptomia por balão segundo o método de Rashkind. Tal correção contribui para o fluxo de sangue no átrio esquerdo e a restauração da circulação sanguínea no grande círculo.
Para a eliminação radical do ADLV, são realizadas intervenções destinadas a criar uma ampla anastomose entre o átrio esquerdo e as veias pulmonares, interrompendo a drenagem patológica das veias pulmonares com outros vasos venosos e eliminando o defeito do septo atrial. Tais intervenções são preferencialmente realizadas em idade precoce. O método de correção radical é selecionado dependendo da variante anatômica do ADLV.
As intervenções corretivas são bastante eficazes, porém, apesar do desenvolvimento da cirurgia cardíaca moderna, o percentual de óbitos ocorridos durante a cirurgia ou no pós-operatório permanece elevado. O resultado da correção radical depende muito da habilidade do cirurgião cardíaco, equipamento técnico, tipo e variante do defeito. Além disso, o maior risco de morte ocorre entre recém-nascidos e crianças pequenas que sofrem de hipertensão pulmonar grave. Na maioria das vezes, essa manifestação do defeito é observada em pacientes com uma variante subcárdica da anomalia.
Os resultados a longo prazo entre as crianças que sobreviveram à correção radical de ADLV são bastante satisfatórios. Apenas alguns pacientes podem desenvolver fraqueza do nó sinusal e hipertensão pulmonar, que são difíceis de responder à terapia medicamentosa.
Previsão
A correção cardiocirúrgica radical do ADLV realizada com sucesso melhora significativamente o prognóstico do desfecho desta cardiopatia. No entanto, o risco de morte durante ou após a cirurgia permanece alto.
Com uma forma total de ADLV e ausência de correção cirúrgica cardíaca de emergência, cerca de 80% das crianças morrem antes de um ano de idade. Se a drenagem venosa pulmonar anormal for parcial, os pacientes podem viver até 20-30 anos. Na ausência de tratamento cirúrgico, esses pacientes morrem de insuficiência cardíaca ou infecções pulmonares.
A drenagem venosa pulmonar anômala é uma cardiopatia congênita rara e sempre requer cirurgia cardíaca. Em alguns casos, a condição de um recém-nascido torna-se crítica devido a distúrbios hemodinâmicos significativos, e tais operações são realizadas em caráter de emergência. Após uma correção cardiocirúrgica radical bem-sucedida, a condição do paciente melhora significativamente e o prognóstico do resultado da doença torna-se favorável.
A drenagem venosa pulmonar anômala (ADPV) é um defeito no qual todas as veias pulmonares (totais) ou individuais (parciais) desembocam no átrio direito ou na veia cava que conduz a ele. A primeira descrição do vício é de J. B. Winslow (1739) e J. G. Wilson (1798). A frequência de ADLV é de 0,5-2% de todas as DCC em combinação com TEA em 10-15%. É provável que um número significativo de casos de ADLV parcial permaneça sem diagnóstico devido à ausência de manifestações clínicas.
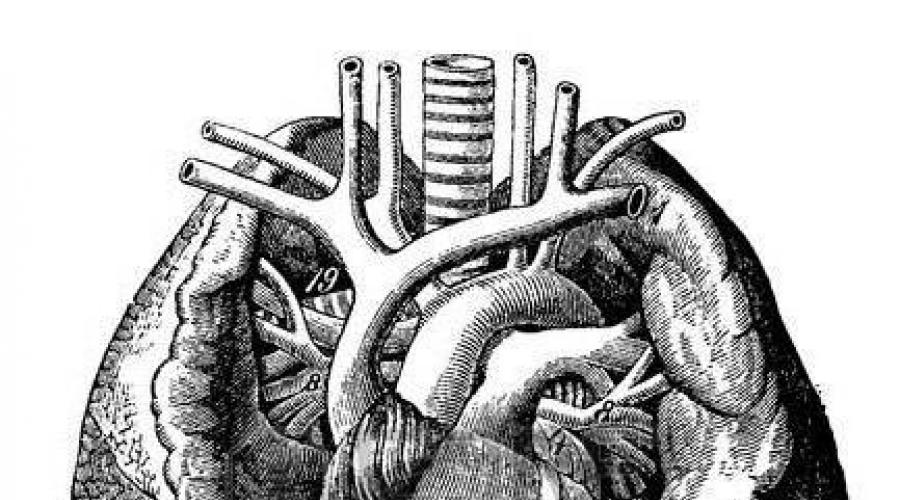
Anatomia, classificação. A drenagem anormal das veias do pulmão direito é mais comum. As seguintes variantes de ADLV podem ser distinguidas (Fig. 12): 1) nível supracardial - pulmonar
a - nível supracárdico: drenagem anômala do JIB na VCS; b - nível cardíaco: drenagem anormal do VP na AD; c - nível infracárdico: drenagem anormal do VE para a VCI; 1 - defeito do septo atrial.
as veias novas desembocam no inominado esquerdo, na veia cava superior ou em um de seus ramos; 2) nível cardíaco - drenagem de todas ou parte das veias pulmonares para a cavidade do átrio direito ou seio coronário; 3) infracardial - parte das veias pulmonares desemboca em um vaso venoso localizado abaixo do coração; 4) misto.
A maioria dos pacientes tem uma CIA secundária concomitante (geralmente um defeito no seio venoso), um forame oval aberto. A autópsia mostra cavidades aumentadas do átrio e ventrículo direitos, artérias pulmonares dilatadas e veia cava; as câmaras esquerdas do coração e a aorta não são alteradas. Geralmente na confluência existem várias bocas das veias pulmonares, às vezes elas se reúnem em um tronco e abrem com uma boca comum. Em 20% dos casos ocorrem outras DCC (CIV, transposição da aorta e artéria pulmonar, tetralogia de Fallot, ventrículo único, estenose pulmonar, dextrocardia).
Hemodinâmica. Com ADLV parcial, os distúrbios hemodinâmicos são semelhantes aos da CIA e são determinados pelo número de veias com drenagem anormal, pelo tamanho do shunt esquerdo-direito e pelo tamanho da CIA associada à DC. Se uma veia pulmonar drena e não há CIA, o defeito permanece assintomático. A pressão na artéria pulmonar com AVD parcial permanece normal por muito tempo, pois o sangue é desviado ao nível dos átrios e não há fator de “pressão de transferência” (do ventrículo esquerdo e aorta para o ventrículo direito e artéria pulmonar, como em VSD e PDA).
Clínica, diagnóstico. Com ADLV parcial, as manifestações clínicas podem estar ausentes por muito tempo ou assemelhar-se às do TEA secundário. Os principais sintomas do defeito não são
sintomas: fadiga, dor no coração, falta de ar durante o exercício, pneumonia repetida, atraso no desenvolvimento físico são possíveis. Com a idade, aparece uma corcunda cardíaca, os limites do embotamento cardíaco se expandem mais para a direita. O quadro auscultatório é semelhante ao da CIA secundária: sopro sistólico não áspero no local de projeção da artéria pulmonar, seccionando
- tons. A insuficiência cardíaca em crianças raramente se desenvolve, geralmente nos casos em que mais de 50% das veias pulmonares são drenadas de forma anormal; em adultos, a deterioração do estado geral aparece mais cedo do que nos casos de TEA isolado.
No exame radiográfico dos órgãos torácicos, o estado do padrão pulmonar é determinado pelo valor da descarga arteriovenosa. Com ADLV parcial, é mais acentuado ao longo do leito arterial se a descarga for superior a 50% do volume minuto da circulação pulmonar. Nos mesmos casos, é possível o abaulamento do tronco da artéria pulmonar e o aumento da pulsação das raízes dos pulmões. A sombra do coração é ampliada devido às seções certas; o átrio direito está dilatado em maior extensão do que na CIA isolada. Com ADLV, sua expansão é visível na veia cava superior. Uma sombra oval das veias pulmonares direitas estendendo-se até a veia cava inferior ao longo da borda direita do coração (padrão "síndrome do sabre" ou "sabre turco") é um sinal confiável de malformação em casos de confluência anormal das veias pulmonares direitas em a veia cava inferior.
Até o momento, não existem critérios ecocardiográficos convincentes para o diagnóstico de drenagem anômala parcial das veias pulmonares. As manifestações indiretas (hemodinâmicas) são: dilatação do ventrículo direito, movimento paradoxal do septo interventricular, tamanhos pequenos do átrio e ventrículo esquerdos, aumento da excursão da válvula tricúspide. Na ecocardiografia 2D, deve-se suspeitar de DVPA parcial quando um defeito septal anatômico é inconsistente com uma manifestação hemodinâmica de shunt arteriovenoso.
Durante a cateterização das cavidades cardíacas, a sonda do átrio direito entra em uma das veias cavas e daí para o campo pulmonar livre. É necessário realizar um estudo faseado da veia cava e do átrio direito, o que permite estabelecer o nível de confluência das veias pulmonares e seu número. Quando a sonda do átrio direito entra no tronco da veia pulmonar, ela tem um curso suave no intervalo entre os orifícios da veia cava e as veias pulmonares, desviando-se para a direita da linha média, ou não passa
medíocre na borda da sombra do átrio direito. Em alguns casos, a sonda pode passar para o tronco da veia pulmonar através da CIA e da cavidade atrial esquerda. A conexão das veias pulmonares com a veia cava é evidenciada pelo aumento da saturação de oxigênio no sangue nos níveis do inominado esquerdo, da veia cava superior ou inferior.
Com drenagem anormal para o átrio direito, nota-se a arterialização do sangue nessa cavidade, o que também é característico de um TEA isolado. A pressão nas câmaras do coração pode ser normal.
A técnica diagnóstica mais valiosa deve ser considerada a angiocardiografia seletiva com a introdução de um agente de contraste no tronco da artéria pulmonar ou seletivamente em seus ramos direito ou esquerdo. Após o enchimento apertado e rápido da artéria pulmonar com um agente de contraste, a fase capilar é visível, então o curso e o local de confluência de todas as veias pulmonares, veias completas, átrio e ventrículo direitos e partes esquerdas do coração são revelados .
Tratamento. Ao corrigir o ADLV parcial, diversas operações são utilizadas dependendo do tipo de defeito (nível de drenagem, tamanho e localização do CIA). Em caso de drenagem anormal de uma parte das veias pulmonares para a veia cava superior ou sua boca, as seguintes táticas são utilizadas: a) criar um túnel com um remendo conectando as bocas das veias pulmonares com drenagem anormal ao átrio esquerdo através de um septo defeito; b) a criação de dois túneis a partir do tronco da veia cava superior para a formação de fluxos sanguíneos separados - sistêmico e pulmonar - e a retirada do sangue das veias pulmonares. diretamente no átrio esquerdo através de uma CIA; c) suturar a borda inferior do defeito à parede posterior da veia cava acima dos orifícios das veias pulmonares com drenagem anormal. Com drenagem anormal das veias pulmonares para a cavidade do átrio direito, a cirurgia é realizada com um remendo. Com uma CIA pequena, ela é expandida para criar um túnel adequado que não interfira no fluxo de sangue das veias pulmonares para o átrio esquerdo. Com drenagem anômala isolada das veias pulmonares para a veia cava superior ou átrio direito, recomenda-se a criação de comunicação interatrial artificial com posterior correção do defeito. Em caso de drenagem anormal das veias pulmonares para a veia cava inferior, utiliza-se um dos métodos acima, ou corta-se o coletor da veia pulmonar e implanta-se no átrio esquerdo. Em caso de drenagem anormal das veias pulmonares esquerdas para a veia vertical esquerda, esta veia é ligada, as veias pulmonares esquerdas são cortadas e implantadas no apêndice atrial esquerdo.
Os resultados da operação são bons. A mortalidade cirúrgica não excede 2-3%. Das complicações, é possível um estreitamento parcial da veia cava superior, drenagem prejudicada das veias pulmonares, danos nos nódulos sinusais ou atrioventriculares.
A vida do paciente só é possível na presença de TEA concomitante. Se não houver CIA, mas houver apenas um canal arterial aberto, os pacientes morrem em idade precoce, pois as partes esquerdas do coração não participam da circulação sanguínea; a morte também ocorre em casos de fechamento prematuro (antes ou logo após o nascimento) do forame oval. Existem muitas variedades deste complexo CHD, portanto, várias classificações foram propostas. A classificação mais simples é R. Darling et al. (1957), eles distinguem quatro tipos de defeito: tipo I - supracárdico - a confluência de todas as veias pulmonares por um tronco comum (coletor) através da veia anômala na veia cava superior, através da veia vertical - no inominado esquerdo, através a veia anômala - na não pareada (47%); Tipo II - cardíaca - confluência de todas as veias pulmonares no átrio direito ou seio coronário (30%);
- tipo - infracárdico - a confluência de todas as veias pulmonares na veia cava portal ou inferior (18%); Tipo IV - misto'- várias combinações desses três tipos (5%) (Fig. 13). Antes de fluir para o coração ou para o vaso que vai para ele, as veias pulmonares se reúnem em uma única câmara - um coletor, que é então conectado à circulação sistêmica em diferentes níveis. Com este tipo de ADLV, há dilatações das câmaras direitas, tronco da artéria pulmonar, enquanto o coração esquerdo costuma estar normal.
Com o AVD total, pode haver causas anatômicas de obstrução na drenagem venosa pulmonar: na maioria das vezes isso se deve ao estreitamento dos orifícios das veias pulmonares com drenagem anormal ou da abertura do coletor, menos frequentemente há pressão externa sobre a veia anormal (na ponto de passagem através do diafragma, entre a artéria pulmonar esquerda e o brônquio principal esquerdo). Em 80% dos casos, a obstrução das veias pulmonares acompanha a variante infra-diafragmática da VAD.
As DAC concomitantes mais comuns no ADLV total são átrio comum, ventrículo único, hipoplasia dos cortes esquerdos, transposição dos grandes vasos (esta combinação cria uma correção funcional do defeito). Em 25-30% dos casos, há malformações extracardíacas do trato gastrointestinal (má rotação do intestino, divertículos, hérnia umbilical), baço (agenesia, múltiplos baços adicionais), urogenital
1- nível supracárdico; 2- nível cardíaco (confluência de todas as veias pulmonares no seio coronário); 3 - nível infracardial.
(cistos renais congênitos, hidronefrose) e sistemas musculoesqueléticos.
Hemodinâmica. Com ADLV total, o sangue de ambos os círculos de circulação sanguínea entra no átrio direito, a magnitude e a direção da descarga são determinadas pela razão das resistências dos grandes e pequenos círculos de circulação sanguínea e relaxamento diastólico dos ventrículos. Uma grande quantidade de sangue arterial entra no átrio direito do coletor, no qual as veias pulmonares fluem. No átrio, o sangue arterial é misturado ao sangue venoso, daqui, através da CIA, parte do sangue (menor) entra no átrio esquerdo e depois na circulação sistêmica; mais sangue é enviado para o pequeno círculo, contribuindo para uma diminuição do coração esquerdo e o desenvolvimento de hipertensão pulmonar. Se um ASD atinge um tamanho significativo e a descarga de sangue na circulação sistêmica é suficiente, o ventrículo esquerdo não é hipoplásico, a hipertensão pulmonar não excede o estágio I-II. e os pacientes podem viver até a idade adulta. Nos casos de obstrução das veias pulmonares, desenvolve-se hipertensão pulmonar venosa; na ausência de involução fisiológica na estrutura dos vasos pulmonares, a hipertensão pulmonar congênita também é possível. Assim, o ADLV total é caracterizado por circulação sanguínea mista com discreta diminuição do conteúdo de oxigênio na circulação sistêmica, sobrecarga do coração direito, hipertensão pulmonar (arterial, venosa, congênita).
Clínica, diagnóstico. O curso clínico do VAD total é determinado pelas características anatômicas e hemodinâmicas do defeito, em particular, o nível de resistência pulmonar geral, o grau de obstrução venosa pulmonar, o tamanho da comunicação interatrial e o estado do miocárdio do ventrículo direito . Os primeiros sinais desse tipo de defeito geralmente aparecem desde os primeiros dias e meses de vida, há manifestações de coração
insuficiência, pneumonia repetida e infecções virais respiratórias agudas, tosse, atraso no desenvolvimento físico. A cianose pode aparecer em qualquer idade, muitas vezes até o final do primeiro ano de vida. Como regra, é insignificante e perceptível apenas ao gritar; cianose pronunciada desde os primeiros dias de vida é característica de obstrução venosa pulmonar. A corcunda cardíaca (principalmente o lado direito) aparece mais cedo do que com DMG1G1 isolado. O tremor sistólico geralmente está ausente. Durante a ausculta, o tom I na área da válvula tricúspide é aumentado (um sinal de aumento do fluxo sanguíneo através dela). O tom II sobre a artéria pulmonar é acentuado, dividido, muitas vezes há um tom III; no segundo espaço intercostal à esquerda, ouve-se sopro sistólico de média intensidade (como na CIA), na borda inferior do esterno à esquerda, é possível um sopro mesodiastólico. Se o coletor flui para a veia cava superior e há obstrução venosa pulmonar, então um sopro sistólico prolongado pode ser ouvido acima da clavícula à direita ou à esquerda.
Em pacientes adultos com ADLV total e hipertensão pulmonar, um tom de ejeção sistólica é ouvido no segundo espaço intercostal à esquerda (ver Capítulo 2). Insuficiência ventricular direita em combinação com sintomas clínicos de TEA e cianose moderada ajuda a suspeitar de DVPA total em crianças. Em pacientes adultos com esse defeito, o quadro clínico é indistinguível do TEA.
No ECG (Fig. 14), um desvio significativo do eixo elétrico do coração para a direita (Z.aAQRS de +90 a +210 °), um aumento no átrio direito (ponta alta Pn.v,), ventrículo direito (em V4R e V| forma qR ', rR', rSR', as ondas R são baixas nas derivações torácicas esquerdas, as ondas S são profundas; esses sinais são especialmente pronunciados na hipertensão pulmonar alta para
ma R a Vi). Pode-se observar a síndrome WPW tipo B. Com o aumento da carga no ventrículo direito, surge um desvio do intervalo ST abaixo da isolinha com ondas T negativas profundas nas derivações II, III, aVF, Vt-Ve.
A fonocardiografia confirma os dados da ausculta e apresenta todos os sinais de um TEA.
Na radiografia com ADLV total, o padrão pulmonar é significativamente aumentado tanto nos canais arteriais quanto venosos, há cardiomegalia moderada ou significativa causada por aumento das câmaras direitas, os cortes esquerdos são de tamanho normal, às vezes uma sombra aumentada do veia cava superior é visível. Para a forma supracárdica do defeito, uma típica sombra cardíaca em forma de oito ou boneco de neve é típica, onde a parte inferior é o próprio coração, e a formação adicional na parte superior direita é um coletor que coleta sangue de todas as artérias pulmonares. veias e se abre na veia cava esquerda ou direita ou veias inominadas (Fig. 15). Estes últimos são dilatados, pois contêm grande volume de fluxo sanguíneo pulmonar. Às vezes, a forma do coração imita uma glândula timo aumentada. Quando as veias pulmonares desembocam no seio coronário ou na veia cava inferior, não há manifestações radiográficas características do defeito.
A M-ecocardiografia não revela um defeito; sinais indiretos são o tamanho relativamente pequeno do átrio esquerdo e ventrículo esquerdo, dilatação do átrio e ventrículo direitos, movimento paradoxal do septo interventricular. Quando as veias pulmonares desembocam no seio coronário, a ecocardiografia bidimensional permite determinar o espaço formado pelo coletor venoso atrás da sombra do coração, o que é confirmado pela administração intravenosa de contraste ultrassonográfico.
A sondagem das cavidades cardíacas com ADLV total revela alta saturação de oxigênio no sangue em todas as partes do coração (90-96%); na artéria pulmonar, a saturação às vezes é a mesma da aorta. A maior saturação é observada no átrio direito ou na veia onde drena o coletor venoso pulmonar. A pressão na artéria pulmonar pode ser aumentada devido a alterações (fetais, escleróticas) nas arteríolas pulmonares, aumento do fluxo sanguíneo pulmonar, obstrução das veias pulmonares (ao mesmo tempo, a pressão capilar pulmonar também aumenta). A pressão nos átrios é igual ou maior à direita do que à esquerda. Nesses casos, pequenas CIAs e um curso leve da doença são observados desde os primeiros dias. Frequentemente mostrado atriosseptostomia urgente (procedimento Rush Kinda) durante o cateterismo.
A introdução de um agente de contraste na artéria pulmonar permite fazer imediatamente o diagnóstico correto (Fig. 16). O agente de contraste encontra-se no coletor da veia pulmonar (CLV), pela veia vertical (VV) entra na veia inominada esquerda (VE), depois na veia cava superior e no átrio direito.
diagnóstico diferencial. Em recém-nascidos e crianças nos primeiros meses de vida, esse defeito é diferenciado de atresia mitral ou aórtica, estenose mitral, coração triatrial, estenose de veia pulmonar, transposição dos grandes vasos, linfangiectasia; em uma idade mais avançada - com VSD.
Curso, tratamento. Cerca de 80% das crianças com VDA total morrem nos primeiros anos de vida, especialmente para malformações infracárdicas (infradiafragmáticas) e outras malformações com obstrução venosa pulmonar. As principais causas de morte em pacientes: insuficiência cardíaca, pneumonia, fechamento prematuro do forame oval. Quanto mais tarde aparecem os primeiros sintomas da doença, melhor o prognóstico. As observações são descritas quando os pacientes viveram até 30 anos; nesses casos, há boa mistura de sangue no nível dos átrios devido ao tamanho suficiente da CIA, não há obstrução venosa, o nível de pressão e a resistência pulmonar geral são baixos e a condição do miocárdio ventricular direito é satisfatório. Na presença de insuficiência cardíaca, é indicada a indicação de glicosídeos cardíacos e diuréticos (ver Capítulo 22). A terapia medicamentosa eficaz permite adiar o tratamento cirúrgico até uma idade mais avançada, pois a correção de um defeito em idade precoce é acompanhada por uma taxa de mortalidade de 35 a 50% [Falkovsky GE et al., 1986; Clarke D. et ai., 1977].
Com ADLV total para o átrio direito (forma cardíaca), a operação é realizada com circulação extracorpórea e hipotermia e consiste em aumentar o defeito interatrial e aplicar um patch de forma a direcionar o fluxo sanguíneo dos orifícios das veias pulmonares através do defeito para o átrio esquerdo. Nos casos em que o coletor drena pelo seio coronário, para realizar a drenagem adequada, é excisada parte do septo localizado entre o seio coronário e a janela oval, incisada a parede posterior superior do seio coronário, o retalho desloca a parede e do seio coronário para o átrio esquerdo. Nos tipos de defeito supracárdicos e infracárdicos, uma anastomose é aplicada entre o coletor da veia pulmonar e o átrio esquerdo [Burakovsky V.I. et al., 1989].
Em caso de hipoplasia grave do ventrículo esquerdo, utiliza-se a correção da CIA com retalho perfurado ou tratamento cirúrgico em dois tempos, incluindo: estágio 1 - anastomose entre o coletor e o átrio esquerdo (permite o desenvolvimento dos cortes esquerdos do coração), estágio II - ligadura do
veia, através da qual as veias pulmonares drenam para a veia cava superior direita.
Mortalidade após cirurgia em crianças menores de 6 anos, segundo
- Galloway et ai (1985) foi de 10%. É maior na forma infracárdica, drenagem das veias pulmonares para a veia cava superior, principalmente para a direita (do que quando desembocam no seio coronário ou diretamente no átrio direito), com hipertensão pulmonar, hipoplasia do ventrículo esquerdo e do átrio esquerdo .
Ao realizar uma operação na infância, os resultados a longo prazo geralmente são bons, pois as alterações escleróticas nos vasos da circulação pulmonar e no miocárdio dos ventrículos do coração não têm tempo de se desenvolver. Se a anastomose apresentar sinais de obstrução funcional, uma segunda operação é indicada.
Neste caso, um tipo de defeito bastante raro, as veias pulmonares não fluem para o átrio esquerdo, mas através de conexões anormais no sistema venoso. Todas as quatro veias pulmonares podem drenar através de um coletor comum para a veia cava superior, para o seio coronário ou para o sistema da veia porta. Muito raramente eles estão conectados diretamente ao átrio direito.
Como com esse defeito, o sangue de ambos os círculos da circulação sanguínea acaba entrando no átrio direito, a circulação sistêmica é mantida pelo sangue que passa pelo forame oval, a menos, é claro, que o canal arterial esteja funcionando ou a pressão nos vasos da circulação pulmonar não excede a dos vasos de grande círculo. As características hemodinâmicas neste tipo de defeito, semelhantes às da HVE sem atresia da válvula mitral ou arterial, não afetam adversamente o desenvolvimento do feto. As partes esquerdas do coração fetal recebem menor quantidade de sangue em relação ao fluxo sanguíneo pulmonar (dependendo do tamanho do forame oval), que não vem pelas veias pulmonares, mas pelo átrio direito. Um ponto importante que determina o curso natural da doença e da sintomatologia é a obstrução concomitante das veias pulmonares, que invariavelmente ocorre quando as veias pulmonares desembocam na circulação sistêmica abaixo do diafragma. A obstrução da veia neste tipo de defeito subdiafragmático é devido ao volume e comprimento do coletor, diminuição do calibre do ducto venoso após o nascimento de uma criança ou estenose do coletor no local de sua fixação à veia sistêmica . Assim, após o nascimento de uma criança, o sangue oxigenado dos pulmões passa pela veia porta e pelo fígado antes de entrar no átrio direito. Pelo contrário, no tipo de defeito supradiafragmático, o sangue oxigenado dos pulmões entra diretamente no seio coronário, ou na veia cava superior através da veia vertical esquerda comum, ou no átrio direito, misturando-se com o sangue venoso sistêmico. . Foi estabelecido que a obstrução venosa de graus variados pode ocorrer com esse tipo de defeito.
Em geral, em crianças com obstrução venosa pulmonar grave, os sintomas aparecem mais precocemente no tipo de defeito subdiafragmático. Nesse caso, a criança pode apresentar edema pulmonar e atelectasia, desenvolvimento de enfisema intersticial e aumento dos ductos linfáticos. Além disso, a hipertrofia da camada média das veias pulmonares e as alterações nas arteríolas pulmonares permanecem inalteradas. Além disso, com obstrução grave das veias pulmonares, as veias brônquicas, que desempenham o papel de colaterais, se expandem. As paredes das pequenas artérias pulmonares engrossam e a massa muscular das arteríolas periféricas aumenta. O coração direito e a artéria pulmonar tendem a estar dilatados, enquanto o coração esquerdo é relativamente pequeno.
Os sintomas da doença em recém-nascidos em que as veias pulmonares não estão obstruídas aparecem mais tarde do que com as veias obstruídas. Ao mesmo tempo, o fluxo sanguíneo pulmonar da criança aumenta acentuadamente. Normalmente, em crianças que desenvolvem sintomas no período pós-natal precoce, a comunicação entre os átrios é pequena, ou seja, é realizada mais pelo forame oval do que por um defeito do septo atrial verdadeiro. A previsão depende do seu tamanho.
A frequência de drenagem venosa pulmonar anômala total é de 2-4% de todos os defeitos cardíacos congênitos que se manifestam no período neonatal, sendo o defeito igualmente comum em meninos e meninas.
Em crianças com esse defeito, o sangue dos vasos dos círculos grandes e pequenos entra no átrio direito e dele - nos vasos dos pulmões e na circulação sistêmica através do orifício no septo atrial e no ventrículo esquerdo. A este respeito, o nível de pO 2 nos vasos de um grande círculo e oxigenação do sangue é inferior ao normal. Seu valor absoluto depende do fluxo sanguíneo pulmonar. Com a obstrução das veias pulmonares, o fluxo sanguíneo para os pulmões é reduzido e pode ocorrer congestão e edema pulmonares, exacerbando a hipoxemia e a cianose. Sem uma operação radical, a criança morre rapidamente. Nesse caso, a hipertensão pulmonar é sempre notada e os sinais de insuficiência cardíaca aparecem precocemente. O tipo de defeito subdiafragmático é fácil de assumir se o nível de pO 2 no sangue da veia umbilical for maior do que na aorta.
Drenagem venosa pulmonar anômala total
A palavra "anômalo" significa "errado". Com este defeito, as veias pulmonares (e são quatro delas), que devem fluir para átrio esquerdo, não caia nele, ou seja. não se conecte com ele. Há muitas opções para sua confluência errada.
Há uma drenagem anômala "parcial" - é quando uma ou duas das quatro veias deságuam no átrio direito (a opção mais comum) e na grande maioria dos casos é combinada com defeitos do septo atrial, e falamos sobre isso no capítulo sobre TEA.
A drenagem venosa pulmonar anômala total ou total (TADPV) é uma questão completamente diferente. Com este defeito, todas as quatro veias pulmonares de ambos os pulmões estão conectadas em um coletor de vasos largo. Esse coletor de sangue arterial oxidado nos pulmões não se funde com o átrio esquerdo, como deveria, mas se conecta ao sistema venoso do corpo, geralmente por meio de uma grande veia. O sangue arterial, assim, contornando o coração, entra nas grandes veias e no átrio direito. Somente aqui, tendo passado pelo defeito do septo interatrial, estará onde deveria estar inicialmente - no átrio esquerdo, e depois faz o trajeto usual pela circulação sistêmica. É difícil imaginar como poderia ser. Mas as crianças com esse defeito nascem a termo, e o coração lida com essa situação por algum tempo. No entanto, este tempo pode ser muito curto.
Em primeiro lugar, a vida de uma criança depende do tamanho da mensagem interatrial - quanto menor, mais difícil é para o sangue arterial chegar ao seu destino na metade esquerda do coração.
Em segundo lugar, nesta metade esquerda do coração, uma parte significativa do sangue é simplesmente venosa, ou seja, não oxidado, e será novamente forçado a formar um grande círculo. Em uma criança, assim, o sangue parcialmente venoso começa a circular nas artérias e ela fica "azul", ou seja, cor da pele, e principalmente - pontas dos dedos e mucosas (lábios, boca) - cianóticas. Isso é cianose, e falaremos sobre suas causas, manifestações e consequências mais adiante.
Com drenagem anômala completa, a cianose pode não ser muito pronunciada, mas está presente e geralmente é perceptível logo após o nascimento.
Na maioria dos casos, a condição de crianças com drenagem venosa pulmonar anômala completa é "crítica" desde o início da vida. Se nada for feito, eles morrerão em poucos dias ou meses.
O tratamento cirúrgico existe, e os resultados hoje são bastante animadores. A operação é bastante complicada, é realizada no coração aberto e consiste na sutura do coletor da veia pulmonar comum ao átrio esquerdo, e o fechamento do orifício do septo atrial com um remendo. Assim, após a operação, a circulação sanguínea normal é restaurada em dois círculos separados.
Às vezes, uma opção de emergência também é aceitável - a expansão do defeito durante a sondagem como o primeiro estágio de salvamento, o que permite atrasar um pouco a intervenção principal.
Não vamos tocar aqui em muitos detalhes relacionados a vários tipos de vício e métodos de corrigi-lo. Mas queremos apenas enfatizar que as crianças com esse defeito precisam especializado imediato ajuda, que hoje é absolutamente real.
Os resultados a longo prazo da operação são bastante bons - afinal, o principal defeito foi eliminado. No entanto, as crianças devem estar sob a supervisão de cardiologistas porque as complicações são possíveis na forma de distúrbios do ritmo ou estreitamento das veias pulmonares nos locais de sutura (isso se deve ao fato de o coração que foi submetido a uma operação tão grande continuar a crescer). E novamente queremos enfatizar: esta criança não é deficiente. Ele deve levar uma vida absolutamente normal, e quanto mais cedo a operação for feita, mais cedo tudo será esquecido.
Pacientes
Comentários
© Copyright 1998 - 2018, Instituição Orçamentária do Estado Federal “N.M. UM. Bakulev" do Ministério da Saúde da Rússia. Todos os direitos reservados.
Doenças → Drenagem anômala total das veias pulmonares
Os materiais postados no site são informações verificadas por especialistas em diversas áreas da medicina e destinam-se exclusivamente a fins educacionais e informativos. O site não fornece aconselhamento e serviços médicos para o diagnóstico e tratamento de doenças. As recomendações e opiniões de especialistas publicadas nas páginas do portal não substituem o atendimento médico qualificado. Possíveis contra-indicações. SEMPRE consulte seu médico.
OBSERVOU UM ERRO NO TEXTO? Selecione-o com o mouse e pressione Ctrl + Enter! OBRIGADO!
Drenagem anômala total de veias pulmonares em recém-nascidos
Todas as veias pulmonares drenam para o coração direito.
Suyaracardial (veias pulmonares drenam na veia vertical ascendente, veia inominada);
Cardíaco (veias pulmonares drenam para o átrio direito, seio coronário);
Infracardial (as veias pulmonares drenam para as veias intra-abdominais).
FISIOPATOLOGIA
Apesar da anatomia diferente da malformação, em termos de fisiopatologia circulatória, ela pode ser classificada em dois tipos dependendo da presença ou ausência de obstrução das veias pulmonares. Importante para a continuação da vida no período pós-natal é a presença de comunicações entre o AD (átrio direito) e o LP (átrio esquerdo) (AMPP, LLC).
A hemodinâmica da variante não obstrutiva da TALV é semelhante à de um TEA maior. O sangue das veias pulmonares entra no AD, depois no VD (ventrículo direito) e, na presença de uma CIA, no AE. Nesse caso, a quantidade de sangue que retorna pela CIA para o AE é determinada pelo tamanho do defeito e pelo grau de extensibilidade da próstata. No entanto, sob quaisquer condições, uma parte muito maior do sangue entra no pâncreas e circula no ICC (circulação pulmonar), causando sobrecarga de volume do pâncreas e hipervolemia acentuada do ICC. A pressão no AE (artéria pulmonar) permanece baixa. Devido ao fato de que a mistura de sangue venoso e arterial proveniente das veias pulmonares ocorre no nível do PP, a saturação no AE e na aorta é idêntica.
Na variante obstrutiva da TALV, a saída de sangue enriquecido com oxigênio das veias pulmonares é difícil. Isso leva ao desenvolvimento de hipertensão venosa pulmonar e, em seguida, a um aumento da pressão no AE e no pâncreas. Cria-se uma situação semelhante à hemodinâmica na estenose da valva mitral. Neste caso, o risco de desenvolver edema pulmonar é alto, porque. a pressão hidrostática nos capilares torna-se muito maior do que a pressão osmótica do sangue. Enquanto a CIA existente fornecer volume suficiente de shunt de sangue da direita para a esquerda, a cavidade pancreática permanece pequena. O lado esquerdo do coração, como no caso da variante não obstrutiva do TALV, permanece "subcarregado" e tem um tamanho relativamente pequeno. O nível de saturação de oxigênio na aorta e no AL é igual, mas seus valores são significativamente menores do que na variante não obstrutiva do defeito. O grau de dessaturação arterial será inversamente proporcional ao volume de fluxo sanguíneo no ICC.
O prognóstico para pacientes com TALV é extremamente desfavorável. Sem correção cirúrgica, dois terços dos pacientes com forma não obstrutiva da doença morrem ao final do primeiro ano de vida.
A causa da morte é muitas vezes pneumonia. Com uma variante obstrutiva, a expectativa de vida é de um mês.
CONSULTÓRIO
uma. Manifestações clínicas da doença:
Variante obstrutiva de TADLV:
Caracterizada por cianose rapidamente progressiva desde o nascimento, que aumenta com a alimentação, associada à compressão das veias pulmonares pelo esôfago;
Falta de ar e sinais de edema pulmonar no período neonatal.
Cianose moderada desde o nascimento;
Atrasado no desenvolvimento físico, infecções broncopulmonares frequentes;
Sinais de insuficiência cardíaca (taquicardia, falta de ar, hepatomegalia).
b. Exame físico:
Variante obstrutiva de TADLV:
Tom II alto na base do coração;
Variante não obstrutiva de TADLV:
O tom II é dividido na base do coração (o componente pulmonar do tom II é acentuado);
Sopro sistólico de intensidade fraca ou moderada (não superior a 3/6) de estenose relativa da valva pulmonar em II m. à esquerda do esterno;
Sopro mesodiastólico de estenose relativa da CT (valva tricúspide) ao longo da borda esquerda do esterno no terço inferior (com fluxo significativo do AD).
DIAGNÓSTICO
- Eletrocardiografia
Variante obstrutiva de TADLV:
hipertrofia do pâncreas (tipo R);
Raramente hipertrofia PP.
Variante não obstrutiva de TADLV:
Hipertrofia do pâncreas de acordo com o tipo de bloqueio da perna direita do feixe de His na derivação Vj (consequência da sobrecarga de volume do pâncreas);
Raramente hipertrofia PP.
O diagnóstico de TALVV é baseado na ausência de todas as veias pulmonares que desembocam no átrio esquerdo em locais típicos.
Dilatação acentuada das partes direitas do coração;
Expansão da artéria pulmonar;
Redução significativa das câmeras esquerdas;
Veia vertical expandida e veia cava superior;
A presença de CIA com descarga direita-esquerda.
TRATAMENTO E OBSERVAÇÃO
- 1. Monitoramento e tratamento de pacientes com TALV não corrigido
A variante obstrutiva do TALV é uma indicação de emergência para cirurgia. Antes de o paciente entrar na clínica de cirurgia cardíaca, as seguintes ações devem ser tomadas:
uma. Infusão IV de preparações de prostaglandina E 1, que manterá o ducto aberto e garantirá a descarga de sangue do AL para a aorta. Isso reduzirá o risco de desenvolver edema pulmonar e aumentará o volume do fluxo sanguíneo no CBC.
dentro. Correção da acidose metabólica.
Variante não obstrutiva de TADLV:
uma. Cuidados intensivos para insuficiência cardíaca (diuréticos, digoxina).
b. Se ocorrer acidose metabólica, ela é corrigida.
Ao estabelecer o diagnóstico de TALV com presença de CIA pequena/restritiva, é realizado o procedimento de Rashkind para aumentar o volume de fluxo sanguíneo no CBC. A indicação para o procedimento é a presença de comunicação restritiva entre os átrios - gradiente de pressão superior a 6 mm Hg.
Indicações para tratamento cirúrgico:
Estabelecer o diagnóstico de TADLV é uma indicação absoluta para a cirurgia.
Contra-indicações para o tratamento cirúrgico:
A presença de contra-indicações absolutas para
Alta resistência venosa dos vasos dos pulmões.
Táticas cirúrgicas
O momento da cirurgia é determinado pela presença ou ausência de obstrução venosa pulmonar.
Na TALV obstrutiva, a intervenção é realizada imediatamente nas primeiras horas após o diagnóstico. Geralmente são as primeiras horas de vida de uma criança.
Na TALV não obstrutiva, o tratamento cirúrgico pode ser postergado e realizado nos primeiros meses de vida.
Técnica cirúrgica
em condições de IR. Aloque um coletor comum LV no local de sua confluência. Normalmente, o coletor é longo o suficiente (mesmo no caso de um dreno infracardíaco) para movê-lo para o átrio esquerdo. O coletor é amplamente aberto, eliminando áreas de estreitamento. Abra bem o átrio esquerdo. Uma ampla anastomose é formada entre o coletor PV e o AE. Elementos adicionais do sistema venoso (veia vertical, etc.) são amarrados.
Complicações específicas do tratamento cirúrgico
Hipertensão pulmonar residual;
Violação do ritmo cardíaco (síndrome de fraqueza do nó sinusal), taquicardia atrial;
Débito cardíaco diminuído devido à obstrução residual da via de saída da VP.
Acompanhamento pós-operatório
- A observação é realizada todos os meses. A duração da observação é determinada individualmente. O monitoramento das complicações tardias da correção cirúrgica do defeito é realizado: estenose da veia pulmonar, distúrbios do ritmo cardíaco.
- Em caso de registro no pós-operatório de NRS, além do exame, recomenda-se o SMEKG a cada 6 meses ou mais frequentemente. Se indicado, é realizada terapia antiarrítmica, RFA ou implante de marcapasso.
- A prevenção da endocardite bacteriana é realizada de acordo com as indicações, se houver estenose das veias pulmonares.
- A admissibilidade da educação física após a correção do defeito. A atividade física é limitada em pacientes com estenose de veia pulmonar.
Se você encontrar um erro, selecione um pedaço de texto e pressione Ctrl+Enter.
Drenagem Anômala Total de Veias Pulmonares (TADLV)
A drenagem anômala total das veias pulmonares é um defeito no qual não há conexão direta entre as veias pulmonares e o átrio esquerdo. As veias pulmonares drenam anormalmente para o átrio direito ou suas tributárias. Quase todos os pacientes têm um forame oval patente ou CIA.
TADLV foi descrito pela primeira vez por Wilson em 1798. Em 1951, Muller realizou a primeira operação bem sucedida. Ela era paliativa. Sem o uso da tecnologia de coração aberto, foi feita uma anastomose entre o coletor da veia pulmonar e a orelha esquerda. Em 1956, Lewis, Varco et al relataram a correção bem-sucedida de um defeito sob condições de hipotermia moderada criada pelo resfriamento da superfície e oclusão temporária do fluxo venoso para o coração. No mesmo ano, Burroughs e Kirklin realizaram a correção de defeitos sob IR. A experiência dos anos subsequentes mostrou que a mortalidade em bebês foi significativamente maior do que em crianças mais velhas. Com o tempo, começaram a surgir relatos na literatura sobre o sucesso das operações em pacientes graves com tipo obstrutivo de TALV. A melhora nos resultados foi associada à introdução de perfusão hipotérmica com redução do volume de fluxo sanguíneo e parada circulatória.
A frequência de ocorrência de TALV, segundo diversos estudos, varia de 0,83 a 2,8%.
Morfogênese
A associação da formação de TADLV com agentes teratogênicos externos conhecidos não foi claramente estabelecida, embora a exposição a chumbo, tintas, solventes e pesticidas possa ser uma causa provável. A natureza genética da TALV não foi identificada, mas um padrão de herança monogênico não é excluído devido aos muitos relatos de casos familiares entre irmãos e irmãs consanguíneos. O gene para esta forma familiar de TADLV está ligado ao cromossomo 4q13-q12. Na mesma região, foi encontrado o gene do receptor do fator de crescimento endotelial vascular, que está associado a uma forma familiar e provavelmente esporádica de TADLW.
O TADLV é combinado com várias síndromes congênitas - asplenia, poliesplenia, síndrome do olho de gato. Com a síndrome do olho de gato, 50% dos pacientes têm CHD como TADLV, tetralogia de Fallot, CIV, ATK.
Anatomia
De acordo com a classificação clínica de Darling et al., dependendo do nível de drenagem, o defeito é representado por quatro opções:
Supracardial. Compõe 50% de todos os TADLV. O coletor venoso pulmonar comum, localizado posteriormente ao átrio esquerdo, drena para o PVVC através das veias vertical esquerda e inominada esquerda.
Intracardíaco. É observada em 20% dos pacientes com TALV. O coletor comum das veias pulmonares drena para o seio coronário, ou desembocam separadamente no átrio direito com quatro bocas.
Infracardial. Ocorre em 20% dos casos. O coletor venoso pulmonar comum drena para a veia porta, ducto venoso, veia hepática e VCI. A veia pulmonar comum, através de uma veia vertical que perfura o diafragma no esôfago, conecta-se às veias porta e à VCI através do ducto venoso ou sinusóides hepáticos.
Misturado. Ocorre em 10% dos pacientes. Este tipo é uma combinação das opções anteriores.
A vida do paciente só é possível na presença de CIA concomitante ou janela oval aberta, que são parte integrante do defeito. Pela mesma razão, persistência do canal arterial em crianças pequenas não é uma anomalia complicada.
Independentemente da localização da conexão anormal das veias pulmonares, o coração em pacientes com TADLV tem características anatômicas comuns: dilatação e hipertrofia do ventrículo direito e átrio direito, dilatação da artéria pulmonar. A metade esquerda do coração é relativamente pouco desenvolvida, especialmente o átrio esquerdo. Seu volume é reduzido para 50% da norma.
Conexão com o átrio direito
A junção geralmente está localizada na parte inferior das costas do átrio direito. A veia pulmonar comum ou duas, três ou quatro veias pulmonares drenam para o átrio direito. O estreitamento das conexões anormais das veias pulmonares é raro. Defeitos cardíacos associados são comuns.
Conexão com o sistema cardinal comum correto
As veias pulmonares de ambos os lados formam um coletor comum localizado atrás do átrio esquerdo. Um único tronco venoso parte do lado direito desse coletor, que sobe, passa em frente à raiz do pulmão direito e flui por trás para a VCS. Em raras ocasiões, ele se conecta com v.azygos. Cirurgiões domésticos encontraram esta variante de TADLV em um paciente. Não foi feito um diagnóstico preciso antes da cirurgia. O exame externo e intracardíaco não revelou a junção do coletor com o coração direito, apesar da VCS estar significativamente expandida. Após criação de anastomose entre o coletor da veia pulmonar e o átrio esquerdo e desmame da CE, houve redução do débito cardíaco, que se manifestou por hipotensão arterial. Com uma tentativa de pinçamento de curta duração da VCS no local de sua confluência com o átrio direito, a pressão arterial aumentou adequadamente. Ficou claro que havia um vazamento de sangue ao longo de um curto trajeto do átrio esquerdo até a VCS. A parede posterior da VCS foi exposta em toda a sua extensão e foi encontrada uma veia ázigos dilatada. Após o curativo, a hemodinâmica normal foi restabelecida.
Conexão com o sistema cardinal comum esquerdo
Veia inominada esquerda.
Nesta, a variante mais comum da TALV, as veias pulmonares de ambos os lados formam um coletor comum atrás do átrio esquerdo. A veia vertical originada do lado esquerdo do coletor geralmente passa anterior à artéria pulmonar esquerda e ao brônquio principal, ascende ao mediastino superior, passa anterior ao arco aórtico e se une à veia inominada esquerda proximal à confluência da jugular esquerda. e veias subclávias. A veia inominada esquerda normalmente se funde com a VCS. Menos comumente, a veia vertical corre entre a artéria pulmonar esquerda e o brônquio principal esquerdo. Essas estruturas o comprimem por fora, causando obstrução do fluxo venoso pulmonar. O tronco venoso que conecta o coletor com a veia inominada esquerda é chamado de veia cava superior esquerda persistente ou veia vertical anômala.
O vaso que conecta o coletor da veia pulmonar diretamente à VCS, contornando a veia inominada, passa entre a artéria pulmonar direita e a traqueia e é comprimido por elas. Uma das variantes da obstrução venosa vertical é o seu estreitamento discreto ou difuso.
A via venosa anormal corre dentro do pericárdio. As veias pulmonares se fundem com um tronco comum, que se une ao seio coronário no sulco atrioventricular. O seio coronário segue então um trajeto normal para o átrio direito, e o orifício fica, como de costume, entre a VCI e a valva tricúspide. As veias do coração drenam normalmente para o seio coronário. Este último é coberto por fibras do átrio esquerdo. O seio coronário tem uma parede comum com o átrio esquerdo em quase toda a sua extensão. Pode haver constrições na junção da veia pulmonar comum com o seio coronário ou dentro dela.
Conexão com o sistema umbilical-vitelino
A junção distal está localizada abaixo do diafragma. As veias pulmonares de ambos os lados formam um coletor comum atrás do átrio esquerdo. O tronco venoso comum parte da parte média do coletor, desce anteriormente ao esôfago e penetra no diafragma pela abertura esofágica. Na maioria das vezes, a veia anômala se une à veia porta na confluência das veias esplênica e mesentérica superior. Menos comumente, o tronco anômalo se conecta ao ducto venoso, a uma das veias hepáticas ou diretamente à veia cava inferior. Nesse tipo de TALV, geralmente ocorre obstrução do retorno venoso pulmonar.
Locais de obstrução anatômica da drenagem venosa pulmonar
Muitos pacientes com tipos supra e intracardíacos de TALV e a maioria dos pacientes com tipo infracardíaco apresentam hipertensão pulmonar devido à obstrução do retorno venoso pulmonar na forma de estreitamento do orifício da veia pulmonar, orifício coletor ou pressão externa na veia anormal. A presença de obstrução no canal venoso pulmonar anormal tem influência decisiva no estado hemodinâmico e nas manifestações clínicas.
Obstrução ao nível do septo interatrial.
Uma pequena comunicação interatrial pode interferir no fluxo de sangue para o átrio esquerdo e, assim, interromper a saída de sangue das veias pulmonares. A expectativa de vida dos pacientes com TALV está obviamente relacionada ao tamanho do TEA. Pacientes com grandes defeitos vivem mais do que aqueles com comunicação atrial restritiva.
O estreitamento das veias pulmonares e suas bocas é especialmente comum no isomerismo atrial. O exame histológico revela espessamento da média e adventícia, bem como proliferação da íntima.
Obstrução de vias venosas anormais.
Uma causa de obstrução é a pressão externa. Assim, na TALV supracárdica, a veia vertical pode passar por trás da artéria pulmonar esquerda e ser comprimida no estreito espaço entre o brônquio, a artéria pulmonar e a persistência do canal arterial ou ligamento. Pode ser comprimido entre a artéria pulmonar direita e a traqueia. Há também um estreitamento interno na passagem típica da veia vertical anterior à artéria pulmonar esquerda. Com TALV infracárdico, a compressão da veia vertical ocorre no esôfago. Nos casos em que a veia vertical está conectada ao ducto venoso, seu estreitamento natural interrompe a saída de sangue das veias pulmonares. Finalmente, quando a veia anômala se une à veia porta ou a uma de suas tributárias, os sinusóides hepáticos comprimem o canal venoso pulmonar, obstruindo o retorno venoso pulmonar.
Atresia da veia pulmonar comum
Raramente, a atresia da veia pulmonar comum é a confluência de todas as veias pulmonares sem conexão com o coração ou veias sistêmicas. A vida útil curta é possível devido às pequenas conexões entre as veias brônquicas e pulmonares. Com esta patologia, a dilatação dos vasos linfáticos pulmonares é pronunciada. A atresia da veia pulmonar comum é a forma mais grave de TALV obstrutiva, apresentando-se com edema pulmonar grave e cianose grave imediatamente após o nascimento. O diagnóstico é estabelecido por ecocardiografia e angiografia. A intervenção cirúrgica é bem sucedida apenas em casos excepcionais. A ECMO é usada antes e após a cirurgia até que a condição dos pulmões melhore.
Defeitos cardíacos associados
TADLV é combinado com muitas DCC, especialmente em pacientes com heterotaxia atrio-abdominal:
uma tétrade com uma válvula pulmonar ausente;
interrupção do arco aórtico;
transposição corrigida e completa dos grandes vasos;
ventrículo com duas saídas;
síndrome de hipoplasia das partes esquerdas do coração;
síndrome do sabre turco.
Microestrutura dos pulmões
O quadro histológico depende da presença ou ausência de obstrução venosa pulmonar. Na ausência de obstrução nas artérias e arteríolas musculares, há hipertrofia acentuada da camada média. O aumento da íntima raramente é visto em bebês, mas é comumente visto em crianças mais velhas e adultos.
A TALV com obstrução é caracterizada por hipertrofia medial nas paredes de canais venosos anormais, veias extrapulmonares e pequenas veias pulmonares. Foi demonstrado que as paredes das artérias e veias pulmonares com drenagem anormal são muito mais espessas do que o normal e com CIV e hipertensão pulmonar. O espessamento da camada média nas artérias e veias é mais pronunciado na presença de obstrução venosa pulmonar. O espessamento da camada muscular nas pequenas artérias pulmonares em TALV correlaciona-se com a pressão na artéria pulmonar. Na mesma pressão na artéria pulmonar, o espessamento medial é sempre mais pronunciado na TALV do que na CIV. A espessura da camada média das veias pulmonares também está intimamente relacionada ao nível de hipertensão arterial pulmonar em pacientes com TAHD. As arteríolas geralmente apresentam proliferação intimal, sendo comum a arterite necrosante.
Os espaços interalveolares são caracterizados por edema e extravasamento de eritrócitos. Os vasos linfáticos subpleurais e interlobulares estão dilatados. Linfangiectasia ocorre em 62% dos pacientes com TALV. É acompanhada de enfisema intersticial, que é uma das causas de mortalidade pós-operatória.
Hemodinâmica
O átrio direito recebe sangue de ambas as circulações. O sangue arterial entra no átrio direito do coletor para o qual as veias pulmonares fluem. No átrio, ele se mistura com o venoso, daqui, pela CIV ou janela oval aberta, parte do sangue entra no átrio esquerdo e depois na circulação sistêmica; a outra parte vai para o pequeno círculo. A natureza da circulação sanguínea depende da distribuição do sangue venoso misto entre os círculos pulmonar e sistêmico. A circulação sanguínea se deve principalmente à magnitude da comunicação interatrial. Se for restritivo, a quantidade de sangue que entra no átrio esquerdo é pequena e o fluxo sanguíneo sistêmico é reduzido. A diminuição do fluxo sanguíneo sistêmico é parcialmente compensada pelo aumento da pressão no átrio direito. Como todas as veias sistêmicas e pulmonares drenam para o átrio direito, a pressão em ambos os poços venosos é aumentada.
Com um forame oval bem aberto ou CIA, há livre circulação de sangue entre os dois átrios. Nessas condições, a distribuição do sangue venoso misto é determinada pela razão das resistências dos grandes e pequenos círculos de circulação sanguínea e a extensibilidade dos ventrículos. Pacientes com obstrução das veias pulmonares desenvolvem hipertensão venosa e, em conexão com isso, hipertensão pulmonar arterial. Na ausência de involução da estrutura fetal das artérias pulmonares, às vezes ocorre hipertensão pulmonar primária.
A TALV é caracterizada por sobrecarga do coração direito e hipertensão pulmonar.
TALV sem obstrução venosa
Ao nascimento, a distribuição do fluxo sanguíneo entre os círculos grandes e pequenos é aproximadamente a mesma, pois a resistência vascular é quase igual. Durante as primeiras semanas de vida, ocorre maturação do leito vascular pulmonar, diminuição da resistência arterial pulmonar e aumento gradual do volume de sangue venoso misto que entra na circulação pulmonar. O fluxo sanguíneo pulmonar é 3-5 vezes maior que o sistêmico. O fluxo sanguíneo sistêmico é normal. Como o pool de sangue venoso misto recebe de 3 a 5 partes de sangue totalmente arterializado para uma parte de sangue venoso sistêmico dessaturado, a saturação de oxigênio do átrio direito pode exceder 90%. Como regra, o sangue no átrio direito se mistura bem, de modo que a saturação do sangue no ventrículo direito, artéria pulmonar, átrio esquerdo, ventrículo esquerdo e aorta é igual à do átrio direito.
Na TALV observam-se dilatação e hipertrofia do ventrículo direito e dilatação da artéria pulmonar.
A pressão da artéria pulmonar em lactentes varia de levemente elevada a sistêmica. Nas poucas crianças que sobrevivem até a adolescência e além, a pressão da artéria pulmonar é ligeiramente elevada. Com o tempo, desenvolve-se hipertorofia medial e proliferação intimal nas arteríolas pulmonares; portanto, observa-se hipertensão mais pronunciada na 3ª e 4ª décadas de vida.
TALV com obstrução venosa pulmonar
O aumento da pressão nos canais venosos pulmonares é transmitido ao leito capilar pulmonar. Quando a pressão hidrostática nos capilares excede a pressão osmótica do sangue, desenvolve-se edema pulmonar. Os mecanismos destinados a prevenir o edema pulmonar incluem aumento do movimento linfático, canais alternativos de derivação venosa pulmonar, permeabilidade reduzida da parede capilar pulmonar e espasmo reflexo das arteríolas. Este último leva à diminuição do fluxo sanguíneo pulmonar, hipertensão pulmonar, aumento da pressão ventricular direita, hipertrofia e insuficiência ventricular direita. Uma diminuição no volume do fluxo sanguíneo pulmonar leva a uma diminuição na proporção de sangue oxigenado no sangue venoso misto e a uma diminuição na saturação nas artérias da circulação sistêmica.
clínica, diagnóstico
O curso clínico do TALVV é determinado pelas características anatômicas e hemodinâmicas do defeito, em particular:
o nível de resistência pulmonar geral;
grau de obstrução venosa pulmonar;
o tamanho da mensagem interatrial;
o estado do miocárdio do ventrículo direito;
a presença de um canal arterial funcionante.
TALV sem obstrução venosa pulmonar
Esses bebês são assintomáticos ao nascer. Logo, metade das crianças desenvolve falta de ar, tosse, dificuldades de alimentação, infecções respiratórias recorrentes e insuficiência cardíaca. Os demais sintomas aparecem no primeiro ano de vida.
A cianose pode aparecer em qualquer idade. Inicialmente, a cianose não é pronunciada e aumenta na presença de insuficiência cardíaca, bem como como resultado do desenvolvimento gradual de alterações secundárias nos vasos pulmonares. No primeiro ano de vida, 75-85% das crianças morrem, a maioria nos primeiros 3 meses. Os bebês estão atrasados no desenvolvimento físico, irritáveis, com choro e esforço, o rosto escurece. Falta de ar, respiração rápida e taquicardia quase sempre ocorrem. A corcunda cardíaca, predominantemente do lado direito, aparece mais cedo do que com um TEA isolado.
O quadro auscultatório é caracterizado pela presença de muitos tons cardíacos. O primeiro tom é alto e distinto, seguido pelo tom de exílio. O segundo tom é amplamente dividido e não muda com os atos de respiração. O componente pulmonar do II tom acentua-se. Quase sempre se ouve um terceiro tom, máximo no topo. Em pacientes mais velhos, um som cardíaco IV é quase sempre ouvido.
Sopros cardíacos podem ocasionalmente estar ausentes. O tremor não é sentido. Um sopro sistólico suave é geralmente ouvido na artéria pulmonar. Muitas vezes, o ruído é bem ouvido no processo xifóide e ao longo da borda inferior do esterno à esquerda. Sopros resultam de fluxo turbulento na via de saída pulmonar e insuficiência da valva tricúspide. Em metade dos casos, um sopro diastólico de aumento do fluxo sanguíneo através da valva tricúspide é ouvido abaixo da borda esquerda do esterno. Ao drenar o coletor na veia inominada esquerda, à direita ou à esquerda na base do coração, pode-se ouvir um sopro sistólico prolongado. Ao contrário dos sopros venosos funcionais, os sopros patológicos não aumentam na diástole e não mudam com mudanças de posição ou pressão nas veias jugulares.
Com insuficiência cardíaca, o fígado está aumentado, há edema periférico. Em algumas crianças que sobreviveram à infância, as falanges terminais dos dedos estão espessadas.
Um achado típico é uma onda P alta e pontiaguda nas derivações II ou direitas, refletindo o aumento do átrio direito. O eixo elétrico é desviado para a direita. Há sempre sinais de hipertrofia ventricular direita, manifestada por alta voltagem dos dentes nas derivações direitas e bloqueio incompleto do bloqueio de ramo direito.
O objetivo da ultrassonografia TALV é confirmar o diagnóstico clínico e localizar o local da drenagem anormal. Os estudos com Doppler podem detectar a presença de obstrução em veias pulmonares individuais e ao longo da veia vertical. Um sintoma comum a todas as formas de TALV é a sobrecarga de volume do ventrículo direito. O ventrículo direito está dilatado. O átrio direito e as artérias pulmonares também estão dilatados, o septo atrial se projeta para a esquerda. Nota-se o movimento paradoxal do septo interventricular.
Além dos sinais de sobrecarga de volume do ventrículo direito, a suspeita de TALV é causada pela incapacidade de traçar o fluxo das veias pulmonares para o átrio esquerdo ao longo do eixo curto paraesternal alto ou supraesternal. A partir disso, além da posição subcostal, pode-se detectar um espaço livre de eco, que é o canal venoso pulmonar comum atrás do átrio esquerdo com as veias pulmonares nele fluindo. Uma vez identificado um coletor venoso comum, deve-se localizar o local de sua drenagem. Na TALVV supracárdica, a veia vertical ascendente drena para uma estrutura venosa sistêmica dilatada, mais comumente para a veia inominada esquerda ou LVVC. O exame com Doppler da veia vertical revela a direção do fluxo sanguíneo para cima e a direção oposta do sangue na veia cava superior.
Com a drenagem do TALV para o seio coronário, este encontra-se dilatado e pode ser detectado ao longo do eixo paraesternal longo ou a partir da posição apical de quatro câmaras.
O exame com Doppler da veia vertical revela a direção do fluxo sanguíneo para cima, em contraste com o movimento oposto na veia cava superior.
Se o coletor venoso pulmonar comum drena para o seio coronário, este encontra-se dilatado e pode ser visto da posição apical de quatro câmaras. O seio coronário dilatado projeta-se anteriormente e para cima no átrio esquerdo porque está localizado no sulco atrioventricular. Se o coletor flui diretamente para o átrio direito, está localizado mais alto do que quando drena para o seio coronário.
Na TALV infradiafragmática, o coletor venoso pulmonar comum drena para o sistema venoso portal, mas pode se conectar às veias hepáticas. As veias pulmonares convergem para um canal comum, muitas vezes pequeno e localizado abaixo do átrio esquerdo e logo acima do diafragma. É visível a partir da posição subcostal. A veia descendente comum situa-se entre a aorta e a VCI. Esses vasos podem ser diferenciados de acordo com a direção e a natureza do fluxo sanguíneo com a ecocardiografia Doppler: na aorta - laminar pulsante na direção do coração, ao longo da veia cava inferior - quase constante em direção ao coração. O fluxo através da veia vertical descendente assemelha-se ao da VCI, mas direcionado para longe do coração.
Para diagnosticar TADLV misto, é necessário utilizar diferentes posições do sensor. O tipo mais comum de drenagem mista é uma combinação de drenagem para o seio coronário e veia inominada esquerda.
O estudo Doppler é o melhor método para avaliar a presença de obstrução das veias pulmonares. O fluxo venoso pulmonar desobstruído é caracterizado por um fluxo quase constante de duas ou três fases, com um curto período de reversão no início da diástole. A obstrução dentro de um dos componentes da via venosa pulmonar se manifesta por corrente monofásica direta de alta velocidade. A obstrução do retorno venoso pulmonar pode ser mascarada se o fluxo sanguíneo pulmonar for reduzido. Pode ser desmascarado pela introdução da prostaglandina E1. O estreitamento ou compressão discreto ao longo do trajeto venoso pulmonar entre estruturas como o ramo da artéria pulmonar e o brônquio pode ser visto diretamente.
Um estudo com Doppler colorido fornece informações sobre a direção e a velocidade média do fluxo através da conexão anômala da veia pulmonar. Isso permite identificar rapidamente a junção do canal venoso pulmonar com a veia sistêmica. A obstrução cria turbulência no fluxo e aparece como um jato colorido em mosaico. Uma vez localizado o local da turbulência, avalie o grau de constrição pela magnitude do pico de velocidade de fluxo espectral na ecocardiografia Doppler.
Em cada tipo de TALV, a possibilidade de malformações concomitantes, como defeitos nas almofadas endocárdicas, atresia pulmonar, ventrículo único, TMA, é cuidadosamente examinada. Erros no diagnóstico de TALV são mais prováveis com anomalias do situs atrial, drenagem venosa pulmonar mista e malformações associadas.
A ecocardiografia pré-natal está sendo cada vez mais usada para diagnosticar defeitos cardíacos em casos de alto risco. A conexão normal das veias pulmonares ao átrio esquerdo pode ser determinada a partir da posição apical de quatro câmaras do coração fetal. Como o fluxo sanguíneo pulmonar fetal é reduzido, a identificação de drenagem venosa pulmonar anormal é difícil. Um sinal indireto de TALV é a expansão do ventrículo direito e da artéria pulmonar, que não é proporcional ao tamanho do ventrículo esquerdo e da aorta, principalmente quando são excluídos outros defeitos que criam o mesmo quadro. Apesar das dificuldades do diagnóstico pré-natal, há relatos isolados na literatura de casos de diagnóstico inequívoco de TALV confirmado após o nascimento. Por outro lado, há casos de TALV que foram detectados pela primeira vez no período neonatal, apesar do ecodiagnóstico pré-natal.
A TALV pode ser diagnosticada por ressonância magnética não invasiva. Este método detecta as veias pulmonares que fluem para a câmara atrás do átrio esquerdo. Pode-se observar que a veia pulmonar comum se conecta à veia inominada esquerda, seio coronário ou átrio direito.
Comum a todos os tipos de TADLV são os sinais de aumento do fluxo sanguíneo pulmonar. O átrio direito e o ventrículo direito estão dilatados e hipertrofiados, o arco da artéria pulmonar abaula. As câmaras das partes esquerdas do coração não estão dilatadas. A junção do coletor venoso pulmonar pode ser refletida na configuração da sombra cardíaca. Com drenagem anormal na veia inominada, a sombra do coração se assemelha a uma figura oito ou a um boneco de neve. A parte superior do "oito" é formada pela veia vertical à esquerda, a veia inominada esquerda por cima e a veia cava superior à direita. Os achados radiográficos característicos geralmente estão ausentes nos primeiros meses de vida e são frequentemente observados em crianças mais velhas e adultos. Com drenagem anormal das veias pulmonares para a veia cava superior, sua dilatação se manifesta pelo abaulamento da borda superior direita da sombra do coração.
As indicações de cateterismo cardíaco são atualmente limitadas devido à alta sensibilidade e especificidade de 99% do diagnóstico ecocardiográfico de TALV. O cateterismo é realizado para esclarecer detalhes importantes que não foram esclarecidos durante o estudo ecocardiográfico:
identificação de defeitos concomitantes;
detecção de locais de conexões anômalas;
localização da obstrução venosa.
O local de uma conexão anormal entre as veias pulmonar e sistêmica pode ser detectado por um “salto” de oxigênio ao nível da veia inominada esquerda, veia cava superior direita ou seio coronário. A drenagem anormal para o átrio direito é identificada pelo aumento da oxigenação no nível do átrio direito, como em outros shunts da esquerda para a direita para o átrio direito. Na TALV, a saturação de oxigênio no átrio direito varia entre 80 e 95%. Aproximadamente o mesmo nível de saturação no ventrículo direito, artéria pulmonar, átrio esquerdo, ventrículo esquerdo e artérias sistêmicas. Com a drenagem anormal do retorno venoso pulmonar para a veia inominada esquerda ou VCS, o sangue misto flui predominantemente para o ventrículo direito e o sangue da veia cava inferior desvia principalmente para o átrio esquerdo, de modo que a saturação sanguínea na artéria pulmonar pode ser maior do que nas artérias sistêmicas.
A pressão no ventrículo direito e na artéria pulmonar varia de discretamente elevada a níveis sistêmicos. A pressão no átrio direito não reflete totalmente a adequação da comunicação interatrial. A presença de pressões iguais em ambos os átrios não é um sinal confiável de um defeito atrial não obstrutivo. Esse fenômeno é explicado pela distensibilidade quase idêntica dos dois ventrículos, de modo que as pressões de enchimento são quase iguais mesmo com comunicação interatrial restritiva. Gradiente de pressão positiva tão baixo quanto 2 mmHg. Arte. ou mais pode indicar um pequeno TEA, mas também pode ocorrer com um relatório maior. A única maneira confiável de estimar o tamanho da mensagem interatrial é medi-la com um cateter balão.
A arteriografia pulmonar seletiva refina significativamente os resultados da ecocardiografia. A passagem do agente de contraste pelas artérias pulmonares e seu aparecimento nos canais venosos pulmonares dá uma ideia clara da anatomia do defeito. Com TALVV, uma veia vertical pode ser vista na veia inominada esquerda, estendendo-se do coletor da veia pulmonar e conectando-se com a veia inominada esquerda. Em seguida, você pode rastrear o fluxo do agente de contraste no SVC.
Com drenagem anormal no seio coronário, o agente de contraste se acumula na veia pulmonar comum e entra no seio coronário na parte posterior do coração. Na projeção anteroposterior, o seio coronário aparece como uma formação ovoide na parte média e inferior do átrio direito. Os detalhes anatômicos do seio coronário são mais bem definidos na vista lateral.
Quando as veias pulmonares drenam diretamente para o átrio direito, as estruturas extracardíacas não são visualizadas e o meio de contraste entra rapidamente no átrio direito.
Se o cateter entrar em uma veia anormal, a saturação sanguínea chega a 100%. É útil a realização de angiografia seletiva, porém, deve-se lembrar que o cateter pode obstruir locais de obstrução venosa pulmonar.
TALV com obstrução da veia pulmonar
A obstrução venosa pulmonar sempre ocorre com TALV infracárdico; em 50% dos casos - com drenagem supracárdica, menos frequentemente - com drenagem para o seio coronário ou para o átrio direito. Independentemente do local da obstrução, o quadro clínico é semelhante. Os sintomas geralmente não aparecem nas primeiras 12 horas de vida, o que permite diferenciar esse defeito da síndrome do desconforto respiratório. Os sintomas de TALV obstrutivo incluem dispneia progressiva, dificuldades de alimentação e insuficiência cardíaca.
As crianças morrem dentro de 2 dias - 4 meses. vida. A drenagem infracárdica é caracterizada por cianose e dispneia, agravada por esforço e deglutição devido ao aumento da pressão intra-abdominal ou compressão da veia pulmonar comum pelo esôfago, como ocorre com a hérnia esofágica.
Apesar dos sintomas graves, os sinais cardiovasculares podem ser mínimos. O coração não está dilatado, o ventrículo direito não está elevado. O segundo tom geralmente é dividido e o componente pulmonar é acentuado. Não há sopro cardíaco, ou é suave, soprando sobre a artéria pulmonar. Os estertores úmidos geralmente são ouvidos nas partes inferiores dos pulmões. A hepatomegalia é quase sempre observada, acompanhada de edema periférico.
O ECG mostra sinais de hipertrofia ventricular direita. Ao contrário do TALV sem obstrução venosa, geralmente não há sinais de aumento do átrio direito.
Com sinais clínicos de TALVV obstrutivo, as veias pulmonares individuais, o coletor da veia pulmonar e sua confluência com as estruturas cardíacas do lado direito devem ser cuidadosamente examinados para localizar o local da constrição. Caracteriza-se por uma natureza de alta velocidade, constante e monofásica do fluxo sanguíneo venoso.
O tamanho da sombra do coração é quase normal. O padrão pulmonar é acentuado e é caracterizado por selos difusos em forma de leque que irradiam das raízes. As bordas da sombra do coração são indistintas. Sinais radiológicos específicos de TALV com obstrução estão ausentes.
A detecção de sangue altamente oxigenado na veia cava superior ou inferior é de grande importância para o diagnóstico. Entretanto, esse fato deve ser interpretado com cautela, pois, por um lado, devido à diminuição do fluxo sanguíneo pulmonar, a saturação do sangue misto pulmonar e venoso sistêmico pode estar baixa, por outro lado, em recém-nascidos com TALV no sistema venoso portal, o sangue da veia umbilical é completamente oxigenado, o que confirma o diagnóstico. A pressão do ventrículo direito é geralmente igual ou maior que a pressão sistêmica. A pressão atrial é normal, em contraste com a alta pressão na vasculatura arterial pulmonar.
A angiografia pulmonar é um método confiável para o diagnóstico de TALV no sistema venoso portal com obstrução venosa. Locais de obstrução também podem ser encontrados em outros locais de drenagem venosa pulmonar anormal. Dado o atraso significativo na passagem do meio de contraste pelo leito vascular pulmonar, ele aparece no sistema venoso pulmonar com atraso de até 12 s após a administração.
Se o cateter entrar em um canal venoso anormal, ele pode passar pela área do estreitamento com ameaça de obstrução significativa ou completa da saída venosa, portanto, a injeção do agente de contraste deve ser feita de forma rápida e manual.
Diagnóstico diferencial
TALV sem obstrução deve ser diferenciado de CIV grande, canal arterial, AOS, DSAV e ventrículo único sem estenose pulmonar. Ao contrário do TALVV, todas essas anomalias apresentam sinais radiográficos e eletrocardiográficos de hipertrofia atrial e ventricular esquerda. Para TALVV, vários sopros cardíacos característicos de defeitos de comparação não são típicos.
Em crianças maiores e adultos, a diferenciação deve ser feita com CIA, átrio comum e PALV. Na TALV, geralmente há cianose pelo menos moderada, o que não ocorre em indivíduos na primeira ou segunda décadas de vida com várias malformações com shunt arteriovenoso no nível atrial.
A TALV com obstrução deve ser diferenciada de outras causas de obstrução venosa pulmonar e de síndrome do coração esquerdo hipoplásico, atresia da valva tricúspide, atresia pulmonar, coarctação, MAT, síndrome do desconforto respiratório e circulação fetal persistente. A cardiomegalia grave geralmente acompanha a atresia valvar e a coarctação da aorta e é uma característica clara não presente na TALV obstrutiva. Um padrão vascular arterial aumentado é característico da MAT e da síndrome do coração esquerdo hipoplásico. A vascularização pulmonar enfraquecida é típica de atresia pulmonar e tricúspide. A obstrução do retorno venoso pulmonar pode ser devido à insuficiência mitral ou insuficiência ventricular esquerda. Essas causas são facilmente diagnosticadas com base em sinais clínicos característicos, em particular hipertrofia e dilatação do ventrículo esquerdo. A ausência de hipertrofia ventricular esquerda é característica da estenose mitral, que também é a causa do fluxo sanguíneo prejudicado dos pulmões. A presença de hipertrofia atrial esquerda indica que a constrição está próxima à valva mitral. Pode ser estenose mitral ou anel estenosante supravalvar do átrio esquerdo, no qual a pressão no átrio esquerdo está aumentada.
Em pacientes com hipertrofia ventricular direita, a TALVV deve ser diferenciada da estenose de veias pulmonares individuais, coração triatrial e atresia da veia pulmonar comum. Em todos esses casos, a pressão no leito arterial dos pulmões está aumentada e a pressão no átrio esquerdo é normal.
A identificação de defeitos obstrutivos é realizada usando angiografia seletiva. Critérios diagnósticos importantes são a detecção de um shunt direito-esquerdo, a duração do preenchimento do átrio esquerdo.
O ecoCG é especialmente informativo na diferenciação de TALV de circulação fetal persistente, síndrome do desconforto respiratório.
fluxo natural
O prognóstico para TALV depende do tamanho da comunicação interatrial, da presença de obstrução venosa pulmonar e da resistência da artéria pulmonar em crianças que têm a sorte de viver por vários anos.
Em uma revisão de Keith et al, incluindo TADP de todos os tipos, 50% das crianças com esse diagnóstico morreram aos 3 meses de idade e 80% no primeiro ano. Os dados de Burroughs e Edwards são semelhantes. Pacientes com comunicação interatrial inadequada têm prognóstico ainda pior. O prognóstico é catastrófico em pacientes com obstrução do canal venoso anormal. Eles geralmente morrem nas primeiras semanas de vida, o filho mais velho viveu 4,5 meses. Alguns pacientes sobrevivem à infância devido ao aumento da resistência arterial pulmonar. Este mecanismo adaptativo pode levar o médico a uma intervenção cirúrgica arriscada. Alterações pronunciadas na íntima das arteríolas pulmonares são detectadas já aos 8 meses de idade.