Normalna prędkość rozprzestrzeniania się płomienia. Wpływ różnych czynników na prędkość płomienia Maksymalna prędkość płomienia spalania propanu
Przeczytaj także
Nad powierzchnią cieczy lub ciała stałego w dowolnej temperaturze znajduje się mieszanina para-powietrze, której ciśnienie w stanie równowagi określa ciśnienie par nasyconych lub ich stężenie. Wraz ze wzrostem temperatury ciśnienie pary nasyconej wzrośnie, ale wykładniczo (równanie Clapeyrona - Clausisa):
gdzie P n „ - ciśnienie pary nasyconej, Pa; Q„ C11 - ciepło parowania, kJ/mol; T - temperatura cieczy, K.
Dla każdej cieczy istnieje zakres temperatur, w którym stężenie par nasyconych nad lustrem (powierzchnia cieczy) będzie znajdować się w obszarze zapłonu, tj. NKPV
W celu wytworzenia LCVV oparów wystarczy podgrzać nie całą ciecz, a jedynie jej warstwę powierzchniową do temperatury równej LTPV.
W obecności źródła zapłonu taka mieszanina będzie zdolna do zapłonu. W praktyce częściej stosuje się pojęcia „temperatury zapłonu” i „temperatury zapłonu”.
Temperatura zapłonu - minimalna temperatura cieczy, przy której nad jej powierzchnią tworzy się koncentracja pary, która może zostać zapalona przez źródło zapłonu, ale szybkość tworzenia się pary jest niewystarczająca do podtrzymania spalania.
Tak więc zarówno w punkcie zapłonu, jak i przy dolnej granicy temperatury zapłonu nad powierzchnią cieczy powstaje dolna granica stężenia zapłonu, jednak w tym ostatnim przypadku DGW tworzą pary nasycone. Dlatego temperatura zapłonu jest zawsze nieco wyższa niż LTLW. Chociaż w punkcie zapłonu następuje krótkotrwały zapłon pary, który nie jest w stanie przekształcić się w stabilne spalanie cieczy, to jednak w pewnych warunkach błysk może spowodować pożar.
Temperatura zapłonu jest przyjmowana jako podstawa klasyfikacji cieczy na palne (ciecze palne) i palne (FL). Ciecze palne obejmują ciecze o temperaturze zapłonu w zamkniętym naczyniu 61 ° C i poniżej, ciecze palne o temperaturze zapłonu powyżej 61 ° C.
Eksperymentalnie określa się temperaturę zapłonu w urządzeniach otwartych i zamkniętych. W naczyniach zamkniętych temperatury zapłonu są zawsze niższe niż w naczyniach otwartych, ponieważ w tym przypadku pary cieczy mają możliwość dyfundowania do atmosfery i wymagana jest wyższa temperatura, aby wytworzyć palne stężenie nad powierzchnią.
W tabeli. 2.4 pokazuje temperaturę zapłonu niektórych cieczy, określoną przez urządzenia typu otwartego i zamkniętego.
Tabela 2.4
Temperatura zapłonu różnych rodzajów cieczy z różnymi metodami oznaczania
Temperatura zapłonu - minimalna temperatura cieczy, przy której po zapłonie par ze źródła zapłonu następuje spalanie stacjonarne.
W cieczach palnych temperatura zapłonu jest wyższa od temperatury zapłonu o 1-5 °, przy czym im niższa temperatura zapłonu, tym mniejsza różnica między temperaturą zapłonu a temperaturą zapłonu.
W przypadku cieczy palnych o wysokiej temperaturze zapłonu różnica między tymi temperaturami sięga 25-35 °. Istnieje korelacja między temperaturą zapłonu w zamkniętym tyglu a dolną granicą temperatury zapłonu, opisaną wzorem
Ta relacja obowiązuje dla Г В(.
Znaczna zależność temperatur zapłonu i zapłonu od warunków doświadczalnych powoduje pewne trudności w stworzeniu metody obliczeniowej szacowania ich wartości. Jedną z najczęstszych z nich jest metoda półempiryczna zaproponowana przez V. I. Blinova:
gdzie G sun - temperatura zapłonu (zapłonu), K; R np - ciśnienie cząstkowe nasyconej pary cieczy w temperaturze zapłonu (zapłonu), Pa; D()- współczynnik dyfuzji par cieczy, s/m 2 ; b- liczba cząsteczek tlenu potrzebnych do całkowitego utlenienia jednej cząsteczki paliwa; W - definicja stałej metody.
Przy obliczaniu temperatury zapłonu w zamkniętym naczyniu zaleca się wziąć W= 28, w naczyniu otwartym W= 45; aby obliczyć temperaturę zapłonu, weź W = 53.
Granice temperatury palności można obliczyć:
Według znanych wartości temperatury wrzenia
gdzie ^n(v)' 7/ip - dolna (górna) temperatura graniczna odpowiednio temperatury zapłonu i wrzenia, °C; k, ja- parametry, których wartości zależą od rodzaju palnej cieczy;
Według znanych wartości granicznych stężeń. Aby to zrobić, najpierw określ stężenie par nasyconych nad powierzchnią cieczy
gdzie (р n to stężenie par nasyconych, %; R n p - prężność pary nasyconej, Pa; R 0 - ciśnienie zewnętrzne (atmosferyczne), Pa.
Ze wzoru (2.41) wynika
Po określeniu ciśnienia pary nasyconej wartością dolnej (górnej) granicy zapłonu znajdujemy temperaturę, w której ciśnienie to jest osiągnięte. Jest to dolna (górna) granica temperatury zapłonu.
Korzystając ze wzoru (2.41), można również rozwiązać problem odwrotny: obliczyć granice stężenia zapłonu ze znanych wartości granic temperatury.
Właściwość płomienia do spontanicznej propagacji obserwuje się nie tylko podczas spalania mieszanin gazów palnych ze środkiem utleniającym, ale także podczas spalania płynów oraz ciała stałe. Pod wpływem lokalnego narażenia na źródło ciepła, na przykład otwarty płomień, ciecz nagrzewa się, szybkość parowania wzrośnie, a gdy powierzchnia cieczy osiągnie temperaturę zapłonu, mieszanina para-powietrze zapali się w miejscu wystawienie na działanie źródła ciepła, powstanie stabilny płomień, który następnie rozprzestrzeni się z określoną prędkością po powierzchni i zimnej części płynów.
Jaka jest siła napędowa propagacji procesu spalania, jaki jest jego mechanizm?
Rozprzestrzenianie się płomienia po powierzchni cieczy następuje w wyniku wymiany ciepła w wyniku promieniowania, konwekcji i przewodzenia ciepła molekularnego ze strefy płomienia na powierzchnię lustra cieczy.
Według współczesnych koncepcji główną siłą napędową rozprzestrzeniania się procesu spalania jest promieniowanie cieplne płomienia. Wiadomo, że płomień o wysokiej temperaturze (powyżej 1000 ° C) może emitować energię cieplną. Zgodnie z prawem Stefana-Boltzmanna intensywność promieniującego strumienia ciepła oddanego przez ogrzane ciało jest określona zależnością
gdzie c ja- intensywność promieniowania cieplnego, kW/m 2 ; 8 0 - stopień czerni ciała (płomień) (e 0 \u003d 0,75-H.0); a = = 5,7 10 11 kJ / (m 2 s K 4) - stała Stefana-Boltzmanna; Г g - temperatura ciała (płomień), K; Г 0 - średnia temperatura, K.
Ciepło promieniujące we wszystkich kierunkach częściowo wnika w obszary powierzchni cieczy, które jeszcze się nie zapaliły, rozgrzewając je. Wraz ze wzrostem temperatury warstwy wierzchniej nad ogrzewanym obszarem nasila się proces parowania cieczy i powstaje mieszanina para-powietrze. Gdy tylko stężenie pary cieczy przekroczy NKVP, zostanie ona zapalona od płomienia. Następnie ten odcinek powierzchni cieczy zaczyna intensywnie nagrzewać sąsiednią część powierzchni cieczy i tak dalej. Szybkość rozprzestrzeniania się płomienia przez ciecz zależy od szybkości nagrzewania powierzchni cieczy przez promieniowanie strumienia ciepła z płomienia, tj. od szybkości tworzenia się palnej mieszaniny para-powietrze nad powierzchnią cieczy, która z kolei zależy od rodzaju cieczy i temperatury początkowej.
Każdy rodzaj cieczy ma swoje własne ciepło parowania i temperaturę zapłonu. Im wyższe ich wartości, tym dłuższy czas potrzebny do jej nagrzania do wytworzenia palnej mieszaniny para-powietrze, tym mniejsza prędkość propagacji płomienia. Wraz ze wzrostem masy cząsteczkowej substancji w tej samej serii homologicznej, prężność par elastyczności maleje, wzrasta ciepło parowania i temperatura zapłonu, a prędkość propagacji płomienia odpowiednio maleje.
Podwyższenie temperatury cieczy zwiększa szybkość rozprzestrzeniania się płomienia, ponieważ skraca się czas potrzebny do podgrzania cieczy do temperatury zapłonu przed strefą spalania.
Podczas błysku prędkość propagacji płomienia wzdłuż lustra cieczy będzie (w kategoriach fizycznych) równa prędkości propagacji płomienia przez mieszaninę para-powietrze kompozycji zbliżonej do LCV, tj. 4-5 cm/s. Wraz ze wzrostem temperatury początkowej cieczy powyżej temperatury zapłonu szybkość propagacji płomienia będzie zależeć (podobnie jak szybkość propagacji płomienia) od składu mieszanki palnej. Rzeczywiście, gdy temperatura cieczy wzrośnie powyżej temperatury zapłonu, stężenie mieszaniny para-powietrze nad powierzchnią lustra wzrośnie od NKVP do 100% (temperatura wrzenia).
Dlatego początkowo, gdy temperatura cieczy wzrasta od temperatury zapłonu do temperatury, w której nad powierzchnią tworzą się pary nasycone, o stężeniu równym stechiometrycznemu (a dokładniej nieco wyższym niż stechiometryczne), szybkość rozprzestrzeniania się płomienia wzrośnie. W naczyniach zamkniętych wraz ze wzrostem temperatury cieczy szybkość propagacji płomienia zaczyna spadać, aż do prędkości odpowiadającej górnej granicy temperatury zapłonu, przy której nie będzie już propagacja płomienia i mieszaniny para-powietrze możliwe ze względu na brak tlenu w mieszaninie para-powietrze nad powierzchnią cieczy. Nad powierzchnią otwartego zbiornika stężenie oparów na różnych poziomach będzie różne: na powierzchni będzie maksymalne i odpowiadać będzie stężeniu oparów nasyconych w danej temperaturze, wraz ze wzrostem odległości od powierzchni stężenie będzie stopniowo zmniejszać się z powodu dyfuzji konwekcyjnej i molekularnej.
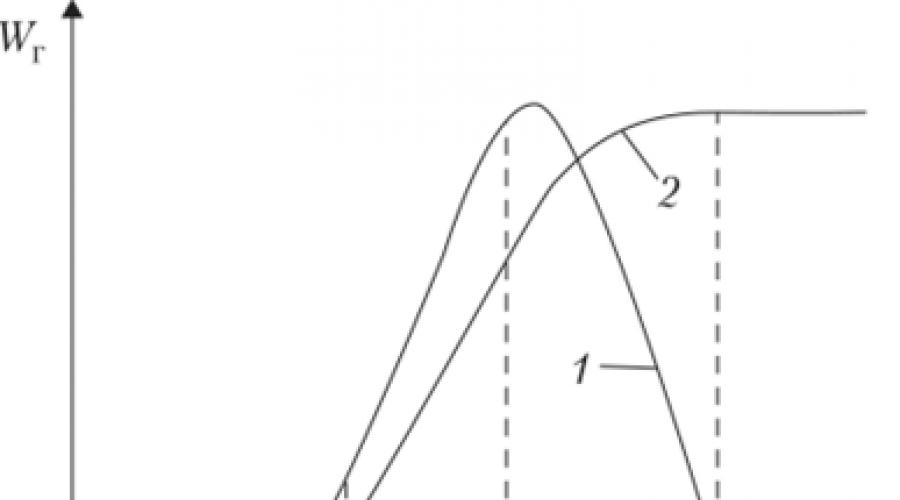
Przy temperaturze cieczy zbliżonej do temperatury zapłonu prędkość propagacji płomienia po powierzchni cieczy będzie równa prędkości jego propagacji przez mieszaninę par w powietrzu przy LIP, tj. 3-4 cm/s. W takim przypadku front płomienia będzie znajdował się w pobliżu powierzchni cieczy. Wraz z dalszym wzrostem temperatury początkowej cieczy prędkość rozchodzenia się płomienia będzie wzrastać podobnie jak normalna prędkość rozchodzenia się płomienia w mieszaninie para-powietrze wraz ze wzrostem jej stężenia. Przy maksymalnej prędkości płomień będzie rozprzestrzeniał się w mieszaninie w stężeniu zbliżonym do stechiometrycznego. W konsekwencji wraz ze wzrostem temperatury początkowej cieczy powyżej G stx szybkość propagacji płomienia pozostanie stała, równa maksymalnej wartości szybkości propagacji spalania w mieszaninie stechiometrycznej lub nieco od niej większa (rys. 2.5). Zatem,

Ryż. 25.
1 - paląca się ciecz w zamkniętym pojemniku; 2 - spalanie cieczy w otwartym zbiorniku ze zmianą początkowej temperatury cieczy w otwartym zbiorniku w szerokim zakresie temperatur (do temperatury wrzenia) prędkość propagacji płomienia będzie wahać się od kilku milimetrów do 3-4 m / s.
Przy maksymalnej prędkości płomień będzie rozprzestrzeniał się w mieszaninie w stężeniu zbliżonym do stechiometrycznego. Wraz ze wzrostem temperatury cieczy powyżej Гstx zwiększy się odległość nad cieczą, przy której powstanie stężenie stechiometryczne, a prędkość propagacji płomienia pozostanie taka sama (patrz rys. 2.5). O tej okoliczności należy zawsze pamiętać, zarówno przy organizacji prac prewencyjnych, jak i przy gaszeniu pożarów, gdy np. może wystąpić niebezpieczeństwo zassania powietrza do zamkniętego pojemnika – jego rozprężenie.
Po zapaleniu cieczy i rozprzestrzenieniu się płomienia, ale jego powierzchnia jest ustalona tryb dyfuzji jego wypalenia, który charakteryzuje się masą właściwą WrM i liniowy W V Jl prędkości.
Prędkość właściwa masy - masa substancji, która wypala się z jednostki powierzchni lustra cieczy w jednostce czasu (kg / (m 2 * s)).
Prędkość liniowa - odległość, na jaką przesuwa się poziom lustra cieczy w jednostce czasu z powodu jego wypalenia (m / s).
Masowe i liniowe szybkości wypalania są ze sobą powiązane poprzez gęstość cieczy p:
Po zapaleniu cieczy temperatura jej powierzchni wzrasta od temperatury zapłonu do wrzenia i tworzy się nagrzana warstwa. W tym okresie tempo wypalania się cieczy stopniowo wzrasta, wysokość płomienia rośnie w zależności od średnicy zbiornika i rodzaju palnej cieczy. Po 1–10 minutach spalania proces stabilizuje się: szybkość wypalania i wymiary płomienia pozostają niezmienione w przyszłości.
Wysokość i kształt płomienia podczas spalania dyfuzyjnego cieczy i gazu podlegają tym samym prawom, gdyż w obu przypadkach o procesie spalania decyduje wzajemna dyfuzja paliwa i utleniacza. Jeżeli jednak podczas spalania dyfuzyjnego gazów prędkość strumienia gazu nie zależy od procesów zachodzących w płomieniu, to podczas spalania cieczy ustala się pewien stopień wypalenia, który zależy zarówno od parametrów termodynamicznych ciecz oraz warunki dyfuzji tlenu z powietrza i pary cieczy.
Pomiędzy strefą spalania a powierzchnią cieczy ustala się pewien transfer ciepła i masy (rys. 2.6). Część strumienia ciepła docierającego do powierzchni cieczy q 0y zużywa się na podgrzanie go do temperatury wrzenia q cn . Dodatkowo ciepły q CT do ogrzewania cieczy pochodzi z pochodni płomienia przez ściany zbiornika w wyniku przewodzenia ciepła. O wystarczająco dużej średnicy q CT można więc zaniedbać q() = K „ n +
To oczywiste, że
gdzie c jest pojemnością cieplną cieczy, kJDkg-K); p jest gęstością cieczy, kg / m3; Wnc- szybkość wzrostu nagrzanej warstwy, m/s; W Jl- liniowy wskaźnik wypalenia, m/s; 0i SP - ciepło parowania, kJ/kg; G kip - temperatura wrzenia cieczy, K.

Ryż. 2.6.
Г () - temperatura początkowa; G kip - temperatura wrzenia;
T g- temperatura spalania; q KUW q Jl - odpowiednio konwekcyjne i promieniste strumienie ciepła; q 0 - strumień ciepła wchodzący na powierzchnię cieczy
Ze wzoru (2.45) wynika, że intensywność przepływu ciepła ze strefy płomienia determinuje pewną szybkość dostarczania paliwa do tej strefy, którego oddziaływanie chemiczne z utleniaczem z kolei wpływa na wartość # 0 . Na tym się składa związek masy oraz wymiana ciepła pomiędzy strefą płomienia a fazą skondensowaną podczas spalania cieczy i ciał stałych.
Oszacowanie udziału ciepła z całkowitego wydzielania ciepła podczas spalania cieczy, które jest zużywane na jego przygotowanie do spalania q 0 , można przeprowadzić w następującej kolejności.
Biorąc za prostotę wrijl= W nx , otrzymujemy
Szybkość wydzielania ciepła na jednostkę powierzchni ciekłego lustra (ciepło właściwe ognia) qll7K) można określić wzorem
gdzie Q H jest najniższą wartością opałową substancji, kJ/kg; P p - współczynnik kompletności spalania.
Następnie biorąc pod uwagę stan (2.44) i dzieląc wyrażenie (2.45) przez wzór (2.46) otrzymujemy
Obliczenia pokazują, że około 2% całkowitego ciepła wydzielanego podczas spalania cieczy jest wydawane na tworzenie i dostarczanie pary cieczy do strefy spalania. Po ustaleniu procesu wypalania temperatura powierzchni cieczy wzrasta do temperatury wrzenia, która następnie pozostaje niezmieniona. To stwierdzenie odnosi się do indywidualnej cieczy. Jeżeli jednak weźmiemy pod uwagę mieszaniny cieczy o różnych temperaturach wrzenia, to najpierw następuje uwalnianie frakcji lekko wrzących, a następnie coraz wyżej wrzących.
Na szybkość wypalania istotny wpływ ma ogrzanie cieczy w głąb w wyniku wymiany ciepła z cieczy ogrzanej przez przepływ promieniujący q0 powierzchnię cieczy na jej głębokość. Ten transfer ciepła jest realizowany przez przewodność cieplna oraz konwencje.
Ogrzewanie cieczy z powodu przewodności cieplnej może być reprezentowane przez wykładniczą zależność postaci
gdzie Tx - temperatura warstwy cieczy na głębokości X, DO; G kip - temperatura powierzchni (temperatura wrzenia), K; k- współczynnik proporcjonalności, m -1 .
Ten rodzaj pola temperatury nazywa się rozkład temperatury pierwszego rodzaju(rys. 2.7).
Konwencja laminarna powstaje w wyniku różnych temperatur cieczy na ściankach zbiornika iw jego środku, a także w wyniku destylacji frakcyjnej w górnej warstwie podczas spalania mieszanki.
Dodatkowa wymiana ciepła z nagrzanych ścianek zbiornika do cieczy prowadzi do nagrzewania się jej warstw przy ściankach do wyższej temperatury niż w środku. Ciecz podgrzana przy ściankach (lub nawet bąbelki pary, jeśli jest podgrzana przy ściankach powyżej temperatury wrzenia) unosi się, co przyczynia się do intensywnego mieszania i szybkiego nagrzewania się płynu na dużej głębokości. Tak zwany warstwa homotermiczna, tych. warstwa o praktycznie stałej temperaturze, której grubość wzrasta podczas spalania. Takie pole temperatury nazywa się rozkład temperatury drugiego rodzaju.

Ryż. 2.7.
1 - rozkład temperatury pierwszego rodzaju; 2 - rozkład temperatury drugiego rodzaju
Powstawanie warstwy homotermicznej jest również możliwe w wyniku destylacji frakcyjnej warstw przypowierzchniowych mieszaniny cieczy o różnych temperaturach wrzenia. W miarę wypalania się takich cieczy warstwa przypowierzchniowa wzbogacana jest w gęstsze, wysokowrzące frakcje, które opadają, przyczyniając się do najbardziej konwekcyjnego ogrzewania cieczy.
Ustalono, że im niższa temperatura wrzenia cieczy (olej napędowy, olej transformatorowy), tym trudniej jest utworzyć warstwę homotermiczną. Kiedy się palą, temperatura ścianek zbiornika rzadko przekracza temperaturę wrzenia. Jednak podczas spalania mokrych wysokowrzących produktów naftowych prawdopodobieństwo powstania warstwy homotermicznej jest dość wysokie. Przy nagrzaniu ścianek zbiornika do temperatury 100°C i wyższej powstają pęcherzyki pary wodnej, które pędząc w górę powodują intensywny ruch całej cieczy i szybkie nagrzewanie się w głąb. Zależność grubości warstwy homotermicznej od czasu palenia opisuje zależność
gdzie X - grubość warstwy homotermicznej w określonym momencie spalania, m; x pr - graniczna grubość warstwy homotermicznej, m; t to czas liczony od początku tworzenia warstwy, s; p - współczynnik, s -1.
Możliwość powstania wystarczająco grubej warstwy homotermicznej podczas spalania mokrych produktów ropopochodnych jest obarczona występowaniem wrzenia i wyrzutu cieczy.
Szybkość wypalania w znacznym stopniu zależy od rodzaju cieczy, temperatury początkowej, wilgotności oraz stężenia tlenu w atmosferze.
Z równania (2.45), biorąc pod uwagę wyrażenie (2.44), można wyznaczyć masowy wskaźnik wypalenia:

Ze wzoru (2.50) wynika, że na szybkość wypalania wpływa intensywność strumienia ciepła od płomienia do lustra cieczy oraz parametry termofizyczne paliwa: temperatura wrzenia, pojemność cieplna i ciepło parowania.
Z tabeli. 2.5 jest oczywiste, że istnieje pewna zależność między szybkością wypalania a kosztami ciepła do podgrzania i odparowania cieczy. Tak więc w serii benzeneksylenogliceroli, wraz ze wzrostem zużycia ciepła do ogrzewania i parowania, szybkość wypalania maleje. Jednak przy przejściu z benzenu do eteru dietylowego koszty ciepła spadają. Ta pozorna rozbieżność wynika z różnicy w intensywności strumieni ciepła dochodzących od płomienia do powierzchni cieczy. Promieniujący strumień jest wystarczająco duży dla płomienia zadymionego benzenu i mały dla stosunkowo przezroczystego płomienia eteru dietylowego. Z reguły stosunek szybkości wypalania najszybciej palących się i najwolniej palących się cieczy jest niewielki i wynosi 3,0-4,5.
Tabela 25
Zależność szybkości wypalania od zużycia ciepła do ogrzewania i parowania
Z wyrażenia (2,50) wynika, że wraz ze wzrostem Г 0 szybkość wypalania wzrasta, ponieważ zmniejszają się koszty ogrzewania cieczy do temperatury wrzenia.
Zawartość wilgoci w mieszaninie zmniejsza szybkość wypalania się cieczy, po pierwsze dzięki dodatkowemu zużyciu ciepła na jej odparowanie, a po drugie w wyniku flegmatyzującego działania pary wodnej w strefie gazowej. Ta ostatnia prowadzi do obniżenia temperatury płomienia, a zatem zgodnie ze wzorem (2.43) zmniejsza się również jego moc promieniowania. Ściśle mówiąc, szybkość spalania mokrej cieczy (cieczy zawierającej wodę) nie jest stała, wzrasta lub maleje w trakcie procesu spalania w zależności od temperatury wrzenia cieczy.
Paliwo mokre można przedstawić jako mieszaninę dwóch cieczy: paliwo + woda, podczas spalania których ich dyspersja frakcyjna. Jeżeli temperatura wrzenia palnej cieczy jest niższa niż temperatura wrzenia wody (100°C), to paliwo wypala się preferencyjnie, mieszankę wzbogaca się w wodę, zmniejsza się szybkość wypalania, a w końcu spalanie ustaje. Jeśli temperatura wrzenia cieczy jest wyższa niż 100 ° C, to przeciwnie, wilgoć najpierw odparowuje, a jej stężenie spada. W rezultacie szybkość spalania cieczy wzrasta, aż do szybkości spalania czystego produktu.
Z reguły wraz ze wzrostem prędkości wiatru wzrasta szybkość wypalania się cieczy. Wiatr intensyfikuje proces mieszania paliwa z utleniaczem, podnosząc tym samym temperaturę płomienia (tabela 2.6) i zbliżając płomień do powierzchni spalania.
Tabela 2.6
Wpływ prędkości wiatru na temperaturę płomienia
Wszystko to zwiększa intensywność przepływu ciepła dostarczanego do ogrzewania i parowania cieczy, a zatem prowadzi do wzrostu szybkości wypalania. Przy wyższych prędkościach wiatru płomień może się zerwać, co prowadzi do ustania spalania. I tak np. kiedy w zbiorniku o średnicy 3 m paliła się nafta traktorowa, wybuchł ogień przy prędkości wiatru 22 m/s.
Większość cieczy nie może palić się w atmosferze zawierającej mniej niż 15% tlenu. Wraz ze wzrostem stężenia tlenu powyżej tej granicy, szybkość spalania wzrasta. W atmosferze znacznie wzbogaconej w tlen spalanie cieczy przebiega z wydzieleniem w płomieniu dużej ilości sadzy i obserwuje się intensywne wrzenie fazy ciekłej. W przypadku cieczy wieloskładnikowych (benzyna, nafta itp.) temperatura powierzchni wzrasta wraz ze wzrostem zawartości tlenu w środowisku.
Wzrost szybkości wypalania i temperatury powierzchni cieczy wraz ze wzrostem stężenia tlenu w atmosferze wynika ze wzrostu emisyjności płomienia w wyniku wzrostu temperatury spalania i dużej zawartości w nim sadzy .
Szybkość wypalania zmienia się również znacząco wraz ze spadkiem poziomu cieczy palnej w zbiorniku: szybkość wypalania maleje, aż do ustania spalania. Ponieważ dopływ tlenu do powietrza z otoczenia wewnątrz zbiornika jest utrudniony, gdy poziom cieczy spada, odległość h np pomiędzy strefą płomienia a powierzchnią spalania (rys. 2.8). Strumień promieniowania do lustra cieczy zmniejsza się, a w konsekwencji zmniejsza się również szybkość wypalania, aż do tłumienia. Przy spalaniu cieczy w zbiornikach o dużej średnicy głębokość graniczna /g pr, przy której następuje tłumienie spalania, jest bardzo duża. Tak więc dla zbiornika o średnicy 5 m jest to 11 m, a przy średnicy Im - około 35 m.

Normalna prędkość rozprzestrzeniania się płomienia to prędkość ruchu czoła płomienia względem niespalonego gazu w kierunku prostopadłym do jego powierzchni.
Wartość normalnej prędkości rozprzestrzeniania się płomienia należy wykorzystać do obliczenia szybkości narastania ciśnienia wybuchu mieszanin gazów i par w zamkniętych, nieszczelnych urządzeniach i pomieszczeniach, średnicy krytycznej (gaszenia) w rozwoju i powstawaniu płomienia ograniczniki, obszar łatwo upuszczanych konstrukcji, membrany bezpieczeństwa i inne urządzenia dekompresyjne; przy opracowywaniu środków zapewniających bezpieczeństwo przeciwpożarowe i przeciwwybuchowe procesów technologicznych zgodnie z wymaganiami GOST 12.1.004 i GOST 12.1.010.
Istotą metody wyznaczania normalnej prędkości propagacji płomienia jest przygotowanie w naczyniu reakcyjnym mieszaniny palnej o znanym składzie, zapalenie jej w środku za pomocą źródła punktowego, rejestracja zmiany ciśnienia w naczyniu w czasie oraz przetworzyć eksperymentalną zależność ciśnienie-czas z wykorzystaniem modelu matematycznego procesu spalania gazu w naczyniu zamkniętym oraz procedur optymalizacyjnych. Model matematyczny pozwala na otrzymanie wyliczonej zależności „ciśnienie-czas”, której optymalizacja według podobnej zależności eksperymentalnej powoduje zmianę prędkości normalnej podczas rozwoju wybuchu dla danej próby.
Normalna szybkość spalania to szybkość, z jaką rozprzestrzenia się czoło płomienia w stosunku do niespalonych reagentów. Szybkość spalania zależy od szeregu właściwości fizykochemicznych odczynników, w szczególności przewodności cieplnej i szybkości reakcji chemicznej, i ma ściśle określoną wartość dla każdego paliwa (w stałych warunkach spalania). W tabeli. 1 pokazuje szybkości spalania (i granice zapłonu) niektórych mieszanin gazowych. Stężenia paliw w mieszankach oznaczono w temperaturze 25°C i normalnym ciśnieniu atmosferycznym. Granice palności, z zaznaczonymi wyjątkami, uzyskano przy propagacji płomienia w rurze o średnicy 0,05 m zamkniętej z obu stron. Współczynniki nadmiaru paliwa określa się jako stosunek objętościowych zawartości paliwa w rzeczywistej mieszance do mieszanki stechiometrycznej (j1) oraz do mieszanki przy maksymalnej szybkości spalania (j2).
Tabela 1
Szybkość spalania mieszanin skondensowanych (utleniacz nieorganiczny + magnez)
| Arkusz |
| Dokument numer. |
| Podpis |
| data |
| Arkusz |
| TGIV 20.05.01.070000.000 PZ |
Jak widać, podczas spalania mieszanin gazowo-powietrznych pod ciśnieniem atmosferycznym ty max mieści się w granicach 0,40-0,55 m/s, oraz - w granicach 0,3-0,6 kg/(m2-s). Tylko dla niektórych związków nienasyconych o niskiej masie cząsteczkowej i wodoru ty max mieści się w granicach 0,8-3,0 m/s i osiąga 1-2 kg/(m2s). Przez powiększenie oraz max badane paliwa w mieszaninach z powietrzem mogą być
ułożyć w następującym rzędzie: benzyna i ciekłe paliwa rakietowe - parafiny i aromaty - tlenek węgla - cykloheksan i cyklopropan - etylen - tlenek propylenu - tlenek etylenu - acetylen - wodór.
| Zmiana |
| Arkusz |
| Dokument numer. |
| Podpis |
| data |
| Arkusz |
| TGIV 20.05.01.070000.000 PZ |
Liniowa szybkość spalania mieszanin tlenowych jest znacznie wyższa niż mieszanin powietrznych (dla wodoru i tlenku węgla - 2-3 razy, a dla metanu - ponad rząd wielkości). Masowe tempo spalania badanych mieszanek tlenowych (z wyjątkiem mieszanki CO + O2) zawiera się w przedziale 3,7–11,6 kg/(m2 s).
W tabeli. Tabela 1 pokazuje (zgodnie z danymi N. A. Silina i D. I. Postovsky'ego) szybkości spalania zagęszczonych mieszanin azotanów i nadchloranów z magnezem. Do sporządzenia mieszanin użyto składników sproszkowanych o uziarnieniu azotanów 150–250 μm, nadchloranów 200–250 μm oraz magnezu 75–105 μm. Mieszanką napełniono tekturowe łuski o średnicy 24-46 mm do współczynnika zagęszczenia 0,86. Próbki wypalano w powietrzu przy normalnym ciśnieniu i temperaturze początkowej.
Z porównania danych w tabeli. 1 i 1,25 wynika z tego, że skondensowane mieszaniny są lepsze od mieszanin gazowych pod względem masy i są od nich gorsze pod względem liniowej szybkości spalania. Szybkość spalania mieszanin z nadchloranami jest mniejsza niż szybkość spalania mieszanin z azotanami, a mieszaniny z azotanami metali alkalicznych spalają się szybciej niż mieszaniny z azotanami metali ziem alkalicznych.
Tabela 2
Granice palności i szybkości spalania mieszanin z powietrzem (I) i tlen (II) w normalnym ciśnieniu i temperaturze pokojowej
| Arkusz |
| Dokument numer. |
| Podpis |
| data |
| Arkusz |
| TGIV 20.05.01.070000.000 PZ |
| Zmiana |
Metody obliczania szybkości wypalania cieczy
| Zmiana |
| Arkusz |
| Dokument numer. |
| Podpis |
| data |
| Arkusz |
| TGIV 20.05.01.070000.000 PZ |
![]() ; (16)
; (16)
gdzie M jest bezwymiarowym wskaźnikiem wypalenia;
 ; (17)
; (17)
M F- masa cząsteczkowa cieczy, kg mol -1 ;
d- charakterystyczna wielkość lustra płonącej cieczy, m. Wyznaczana jest jako pierwiastek kwadratowy z powierzchni spalania; jeżeli obszar spalania ma kształt koła, to charakterystyczny rozmiar jest równy jego średnicy. Przy obliczaniu szybkości spalania turbulentnego można przyjąć d= 10 m;
T do to temperatura wrzenia cieczy, K.
Procedura obliczeniowa jest następująca.
Tryb spalania zależy od wartości kryterium Galileusza Ga, obliczone według wzoru
gdzie g- przyspieszenie swobodnego spadania, m·s -2 .
W zależności od trybu spalania obliczana jest bezwymiarowa szybkość wypalania M. Dla trybu spalania laminarnego:
Dla przejściowego trybu spalania:
Jeśli następnie ![]() , (20)
, (20)
jeśli , to , (21)
Dla reżimu spalania turbulentnego:
; ![]() , (22)
, (22)
M0- masa cząsteczkowa tlenu, kg mol -1 ;
n 0- współczynnik stechiometryczny tlenu w reakcji spalania;
n F- współczynnik stechiometryczny cieczy w reakcji spalania.
B- bezwymiarowy parametr charakteryzujący intensywność wnikania masy, obliczony ze wzoru
![]() , (23)
, (23)
gdzie Q- wartość opałowa cieczy, kJ·kg -1 ;
| Zmiana |
| Arkusz |
| Dokument numer. |
| Podpis |
| data |
| Arkusz |
| TGIV 20.05.01.070000.000 PZ |
c- izobaryczna pojemność cieplna produktów spalania (przyjęta jako równa pojemności cieplnej powietrza c = 1), kJ·kg -1 ·K -1 ;
T0- temperatura otoczenia, przyjęta jako 293 K;
H- ciepło parowania cieczy w temperaturze wrzenia, kJ·kg -1 ;
c e to średnia izobaryczna pojemność cieplna cieczy w zakresie od T0 zanim do.
Jeżeli znana jest lepkość kinematyczna pary lub masa cząsteczkowa i temperatura wrzenia badanej cieczy, wówczas szybkość spalania turbulentnego oblicza się na podstawie danych doświadczalnych według wzoru
gdzie ja- eksperymentalna wartość szybkości wypalania w stanie nieustalonym, kg·m -2 ·s -1 ;
d ja- średnica palnika, w którym uzyskuje się wartość ja, m. Zaleca się stosowanie latarki o średnicy 30 mm. Jeżeli w palniku o średnicy 30 mm obserwuje się reżim spalania laminarnego, należy zastosować palnik o większej średnicy.
3. ROZPATRYWANIE PŁOMIENIA W MIESZANKACH GAZOWYCH
Szybkość rozprzestrzeniania się płomienia podczas spalania substancji stałych, ciekłych i gazowych ma praktyczne znaczenie z punktu widzenia zapobiegania pożarom i wybuchom. Rozważ prędkość rozprzestrzeniania się płomienia w mieszaninach palnych gazów i par z powietrzem. Znając tę prędkość można wyznaczyć bezpieczną prędkość przepływu gazu-powietrza w rurociągu, kopalni, instalacji wentylacyjnej i innych systemach wybuchowych.
3.1. PRĘDKOŚĆ PŁOMIENIA
Jako przykład na ryc. 3.1 przedstawia schemat wentylacji wyciągowej w kopalni węgla kamiennego. Z wyrobisk kopalni 1 rurociągiem 2 usuwana jest pylista mieszanina powietrza i pyłu węglowego, aw niektórych przypadkach metan uwalniany w pokładach węgla. Gdy wybuchnie pożar, front płomienia 3 rozprzestrzeni się w kierunku zaspy 1. Jeśli prędkość mieszanki palnejw będzie mniejsza niż prędkość propagacji frontu płomieniaoraz w stosunku do ścian rury płomień rozprzestrzeni się w kopalni i doprowadzi do wybuchu. Dlatego do normalnej pracy systemu wentylacyjnego konieczne jest spełnienie warunku
w>u.
Szybkość usuwania mieszaniny wybuchowej musi być większa niż szybkość propagacji frontu płomienia. Zapobiegnie to przedostawaniu się płomieni do wyrobisk szybowych.
Ryż. 3.1. Schemat propagacji płomienia w kopalni:
1 - mój; 2 - rurociąg; 3 - front płomienia
Teoria propagacji płomienia rozwinięta w pracach Ya.B. Zeldovich i D.A. Frank-Kamenetsky opiera się na równaniach przewodnictwa cieplnego, dyfuzji i kinetyki chemicznej. Zapłon mieszanki palnej zawsze zaczyna się w jednym punkcie i rozciąga się na całą objętość zajmowaną przez mieszankę palną. Rozważ jednowymiarowy przypadek - rurkę wypełnioną palną mieszanką (ryc. 3.2).
Jeśli mieszanina zostanie zapalona z jednego końca rury, wąski front płomienia rozprzestrzeni się wzdłuż rury, oddzielając produkty spalania (za frontem płomienia) od świeżej mieszanki palnej. Czoło płomienia ma formę czapeczki lub stożka z częścią wypukłą zwróconą w kierunku ruchu płomienia. Front płomienia to cienka warstwa gazowa o szerokości (10 -4 ÷ 10 6) m. W tej warstwie, zwanej strefą spalania, zachodzą chemiczne reakcje spalania. Temperatura czoła płomienia, w zależności od składu mieszanki, wynosi T= (1500 ÷ 3000) K. Uwolnione ciepło spalania jest zużywane na ogrzewanie produktów spalania świeżej mieszanki palnej i ścian rur w wyniku procesów przewodzenia ciepła i promieniowania.

Ryż. 3.2. Schemat propagacji frontu płomienia w rurze
Gdy czoło płomienia porusza się w rurze, w mieszaninie palnej powstają fale sprężania, które wywołują ruchy wirowe. Zawirowania gazu uginają front płomienia, nie zmieniając jego grubości i charakteru zachodzących w nim procesów. Na jednostkowej powierzchni czoła płomienia zawsze pali się taka sama ilość substancji w jednostce czasu. ![]() . Wartość jest stała dla każdej mieszanki palnej i nazywana jest szybkością spalania masowego .
Znajomość obszaru frontu płomieniaS, możesz obliczyć masę substancji M, palny w całym czole spalania w jednostce czasu:
. Wartość jest stała dla każdej mieszanki palnej i nazywana jest szybkością spalania masowego .
Znajomość obszaru frontu płomieniaS, możesz obliczyć masę substancji M, palny w całym czole spalania w jednostce czasu:
Każdy element frontu płomienia dSporusza się względem świeżej mieszanki zawsze w kierunku normalnym do czoła płomienia w danym punkcie (rys. 3.2), a prędkość tego ruchu:
gdzie jest gęstość świeżej mieszanki palnej.
Wartość nazywana jest normalną prędkością propagacji płomienia i ma wymiar m/s. Jest to stała wartość procesu spalania danej mieszaniny i nie zależy od warunków hydrodynamicznych towarzyszących procesowi spalania. Normalna prędkość propagacji płomienia jest zawsze mniejsza niż prędkość obserwowana oraz, czyli prędkość frontu spalania względem ścianek rur:
ty nie< u .
Jeżeli front płomienia jest płaski i skierowany prostopadle do osi rury, to w tym przypadku obserwowana i normalna prędkość propagacji płomienia będzie taka sama
u n = u .
Powierzchnia wypukłego czoła płomieniaS problemzawsze większa niż powierzchnia frontu płaskiegoS pl, Dlatego
> 1.
Normalna prędkość płomieniaty niedla każdej mieszanki palnej zależy od domieszki gazów obojętnych, temperatury mieszanki, wilgotności i innych czynników. W szczególności, wstępne podgrzewanie gazu palnego zwiększa szybkość rozprzestrzeniania się płomienia. Można wykazać, że prędkość propagacji płomieniaty niejest proporcjonalna do kwadratu temperatury bezwzględnej mieszaniny:
u n .= const T 2.
Na ryc. 3.3 pokazuje zależność prędkości propagacji płomienia w palnej mieszaninie „powietrze – tlenek węgla” w zależności od stężenia CO. Jak wynika z powyższych wykresów, prędkość rozprzestrzeniania się płomienia wzrasta wraz ze wzrostem temperatury mieszaniny. Dla każdej wartości temperatury prędkość propagacji płomienia ma maksimum w obszarze stężenia tlenku węgla CO równe ~ 40%.
Pojemność cieplna gazu obojętnego wpływa na szybkość rozprzestrzeniania się płomienia. Im większa pojemność cieplna gazu obojętnego, tym bardziej obniża on temperaturę spalania i tym bardziej zmniejsza prędkość propagacji płomienia. Tak więc, jeśli mieszanina metanu z powietrzem zostanie rozcieńczona dwutlenkiem węgla, prędkość rozprzestrzeniania się płomienia może zmniejszyć się 2-3 razy. Na szybkość rozprzestrzeniania się płomienia w mieszaninach tlenku węgla z powietrzem duży wpływ ma wilgoć zawarta w mieszaninie, obecność cząstek sadzy oraz zanieczyszczenia gazów obojętnych.

Ryż. 3.3. Zależność prędkości propagacji płomienia
o stężeniu tlenku węgla w mieszaninie
Podział strefy przemian chemicznych w otwartym układzie palnym
Spalanie rozpoczyna się od zapalenia mieszanki palnej w lokalnej objętości układu palnego, a następnie rozprzestrzenia się w kierunku poruszającej się mieszanki. Strefa spalania, w której zachodzą reakcje chemiczne redoks widoczne dla obserwatora, nazywana jest płomieniem. Jako front płomienia służy powierzchnia oddzielająca płomień od niepłonącej się jeszcze mieszanki. Charakter rozprzestrzeniania się płomienia zależy od wielu procesów, ale decydującym czynnikiem jest proces nagrzewania mieszanki palnej. W zależności od sposobu podgrzewania mieszaniny palnej do temperatury zapłonu rozróżnia się propagację płomienia normalną, turbulentną i detonacyjną.
Podczas spalania w układzie palnym z mieszanką poruszającą się laminarnie obserwuje się normalne rozprzestrzenianie się płomienia. Podczas normalnego rozprzestrzeniania się płomienia energia cieplna jest przekazywana z warstwy palącej się do zimnej głównie przez przewodnictwo cieplne, a także przez dyfuzję molekularną. Przewodność cieplna w gazach charakteryzuje się małą intensywnością, dzięki czemu prędkość normalnego rozprzestrzeniania się płomienia jest niewielka.
Podczas turbulentnego ruchu mieszanki palnej przenoszenie energii cieplnej z warstwy palącej się do zimnej następuje głównie poprzez dyfuzję molową, a także przewodność cieplną. Transfer molowy jest proporcjonalny do skali turbulencji, która jest określona przez prędkość mieszaniny. Szybkość burzliwego propagacji płomienia zależy od właściwości mieszaniny i dynamiki przepływu gazu.
Rozprzestrzenianie się płomienia w palnej mieszaninie ze strefy spalania do zimnych warstw poprzez procesy molekularne i molowe nazywamy deflagracją.
Procesom fizykochemicznym spalania towarzyszy wzrost temperatury i ciśnienia w płomieniu. W układach palnych w określonych warunkach mogą powstawać strefy wysokiego ciśnienia, które mogą ściskać sąsiednie warstwy, podgrzewając je do stanu zapłonu. Rozprzestrzenianie się płomienia poprzez gwałtowne sprężanie zimnej mieszanki do temperatury zapłonu nazywa się detonacją i jest zawsze wybuchowe.
W układach palnych może wystąpić spalanie wibracyjne, w którym front płomienia porusza się z prędkością zmienną zarówno pod względem wielkości, jak i kierunku.
Prędkość propagacji frontu spalania w mieszance poruszającej się laminarnie lub nieruchomej nazywana jest normalną lub podstawową prędkością propagacji płomienia. Wartość liczbowa normalnej prędkości jest określona przez prędkość mieszanki, która jeszcze się nie zapaliła, zwykle skierowana w stronę frontu spalania.
Wartość u n dla płaskiego frontu spalania można wyznaczyć z warunku równowagi dynamicznej między szybkością nagrzewania mieszaniny przez przewodność cieplną do temperatury zapłonu a szybkością reakcji chemicznej. Rezultatem jest następująca formuła
gdzie l jest współczynnikiem przewodzenia ciepła mieszaniny gazów, ср jest współczynnikiem pojemności cieplnej mieszaniny przy stałym ciśnieniu, Тin jest temperaturą początkową mieszaniny, Та jest adiabatyczną temperaturą spalania, Arr jest kryterium Arrheniusa, k 0 jest współczynnik prawa Arrheniusa.
Normalną prędkość można wyznaczyć doświadczalnie z prędkości czoła w rurze z nieruchomą mieszanką lub z wysokości stożka spalania w palniku Bunsena. Palnik Bunsena to palnik laboratoryjny z częściowym wstępnym mieszaniem gazu i powietrza. Na wylocie palnika powstaje płomień z frontem spalania w postaci stożka o regularnym kształcie (rys.).
Rys.7. Front spalania w palniku Bunsena
Przy stabilnym położeniu frontu spalania prędkość propagacji płomienia u n jest równoważona przez składnik W n prostopadły do powierzchni stożka spalania oraz prędkość mieszaniny gaz-powietrze W, tj.
gdzie j jest kątem między wektorem prędkości mieszaniny gaz-powietrze a wektorem jej składowej normalnej do powierzchni stożka spalania.
Wartość prędkości ruchu mieszaniny gaz-powietrze na wylocie dyszy ze stożkiem spalania o regularnym kształcie określa wzór
gdzie d 0 jest średnicą dyszy palnika, V jest natężeniem przepływu mieszaniny gaz-powietrze przez palnik.
Wartość cos j można wyrazić w postaci wysokości stożka spalania
Biorąc pod uwagę fakt, że powierzchnia spalania jest boczną powierzchnią zwykłego stożka
określana jest wartość normalnej prędkości
Na wartość normalnej prędkości rozprzestrzeniania się płomienia mają wpływ:
1. Temperatura początkowa mieszaniny. W niskich temperaturach un jest wprost proporcjonalna do kwadratu temperatury bezwzględnej mieszaniny wchodzącej do spalania. W temperaturze powyżej temperatury zapłonu pojęcie normalnej prędkości traci sens, ponieważ mieszanina staje się zdolna do samozapłonu.
2. Temperatura ścianek kanału pod warunkiem, że płomień rozprzestrzenia się wewnątrz tego kanału. Zimne ściany przerywają reakcje łańcuchowe i spowalniają rozprzestrzenianie się płomienia.
3. Średnica kanału. Dla każdej palnej mieszanki istnieje wartość krytyczna średnicy d cr, od której niemożliwe jest rozprzestrzenianie się płomienia w kanale. Wartość średnicy krytycznej można określić wzorem
gdzie cm jest dyfuzyjnością cieplną mieszaniny.
4. Ciśnienie. Wraz ze wzrostem ciśnienia u n maleje.
5. Skład mieszanki. Dla mieszaniny o składzie zbliżonym do stechiometrycznego normalna prędkość ma wartość maksymalną. Ponadto istnieją dolne i górne granice w zakresie stężenia paliwa, powyżej których płomień nie może się rozprzestrzeniać.
Smary
Głównym celem w rozwoju smarów przyjaznych dla środowiska jest stworzenie produktu o wysokiej biodegradowalności i niskiej ekotoksyczności. W rozwiniętych krajach Zachodu
Obecnie firmy publiczne i prywatne zaczynają tworzyć rynek smarów przyjaznych dla środowiska. Większość badań koncentruje się na składzie chemicznym produktu i ocenie jego biodegradowalności. Tworząc przyjazne dla środowiska środki smarne bierze się pod uwagę dwa główne obszary: produkcję olejów bazowych, których chemiczny charakter determinuje charakter oddziaływania na środowisko oraz syntezę nowych dodatków, które są przyjazne dla środowiska, biodegradowalne i skuteczne.
Obecnie, a prawdopodobnie w przyszłości, szczególne znaczenie mają trzy grupy olejów bazowych pochodzących z różnych źródeł wsadowych: oleje naftowe z hydrokrakingu (HA), polialfaolefiny (PAO) oraz estry, które ulegają szybkiej biodegradacji w środowisku. Ogromne znaczenie przez nieskończenie długi czas bez wątpienia pozostaną bazowe oleje naftowe o tradycyjnych schematach przepływowych, zwłaszcza biorąc pod uwagę czynnik, jakim są smary otrzymywane na bazie PAO. estry polialkoholi, glikole polialkilenowe i diestry kosztują 2-10 razy więcej niż produkty naftowe. Zwiększona biodegradowalność nie stanowi zachęty do przezwyciężania różnic cenowych.
Wysokie właściwości użytkowe i przyjazność dla środowiska olejów mineralnych zapewnia zestaw pewnych cech. Przede wszystkim jest to ich wąski ułamkowy i korzystny skład chemiczny grupowy z minimalną zawartością związków siarki i azotu w olejach bazowych. Dobór surowców, sortowanie olejów wykorzystywanych do produkcji olejów o wysokim indeksie oraz ich odrębna obróbka mają ogromne znaczenie. W uzyskiwaniu bazowych olejów mineralnych spełniających wymagania środowiskowe ważną rolę odgrywa selektywne oczyszczanie,
znacząca rakotwórczość produktu. Obecnie w USA i Kanadzie ponad 70% olejów bazowych pozyskiwane jest poprzez selektywną rafinację. Zastosowanie tak nowoczesnych procesów jak hydrokraking, hydroodparafinowanie, hydroizomeryzacja otwiera szerokie możliwości. Technologie te zostały szczegółowo opisane w pracy. Zastosowanie procesów hydrokatalitycznych w połączeniu z tradycyjnymi metodami rafinacji surowców olejowych selektywnymi rozpuszczalnikami poprawia wydajność i właściwości środowiskowe olejów bazowych.
W tabeli. W tabeli 1.4 przedstawiono dane porównawcze dotyczące składu chemicznego olejów bazowych otrzymanych metodą selektywnej rafinacji i hydrorafinacji. Ten ostatni znacznie obniża zawartość aren, siarki i azotu w olejach.
Tabela 14
Wpływ hydrorafinacji na skład chemiczny
oleje bazowe
Wprowadzenie do produkcji bazowych olejów mineralnych procesów hydrokrakingu i hydroizomeryzacji umożliwia otrzymanie produktów o zwiększonej biodegradowalności i wolnych od arenów. Oleje do hydrokrakingu, zgodnie z wynikami uzyskanymi nowoczesnymi metodami badawczymi, są nietoksyczne, praktyczny brak w nich arenów wskazuje na bardzo niską rakotwórczość i znikome prawdopodobieństwo jego wzrostu poprzez tworzenie i akumulację aren wielopierścieniowych podczas eksploatacji; brak aren i przewagi
Podawanie izoparafin zapewnia dość wysoką biodegradowalność.
Oleje bazowe z hydrokrakingu są produkowane w USA od końca 1996 roku. . Do uruchomienia została przygotowana instalacja w Finlandii.
W Rosji VNIINP wraz z centrum naukowo-technicznym OAO LUKOIL i AO LUKOIL - Volgogradneftepererabotka prowadzą badania nad organizacją produkcji szeregu rzadkich olejów i baz z wykorzystaniem technologii uwodorniania, w szczególności oleju lotniczego MS-8 i AMG lotniczy płyn hydrauliczny -dziesięć.
W porównaniu z olejami mineralnymi, oleje syntetyczne w niektórych przypadkach mają lepsze właściwości środowiskowe. Do najważniejszych klas olejów syntetycznych z punktu widzenia bezpieczeństwa środowiskowego należą oleje wytwarzane na bazie estrów syntetycznych, polialfao-lefin i polibutenów. Są nietoksyczne, nierakotwórcze, charakteryzują się niską emisją szkodliwych substancji.
Oleje syntetyczne na bazie estrów z dodatkami są szeroko stosowane w turbinowych silnikach gazowych samolotów cywilnych i wojskowych od lat 60. XX wieku. W CIAM wraz z VNIINP i 25. Państwowym Instytutem Badawczym Ministerstwa Obrony Federacji Rosyjskiej trwają prace nad stworzeniem wysoce stabilnego termicznie (do 240 ° C) oleju estrowego przy użyciu skutecznych kompozycji dodatków, które nie są gorszej jakości do najlepszych olejów zagranicznych. Analiza informacji naukowych, technicznych i patentowych dotyczących olejów do lotniczych turbinowych silników spalinowych wskazuje, że poliestry pozostają główną klasą związków stosowanych jako materiały bazowe [PO]. Jednak sytuacja zmienia się wraz z następną generacją silników lotniczych, ponieważ ulepszenia konstrukcyjne i potrzeba zmniejszenia zużycia paliwa prowadzą do wzrostu ciśnienia, temperatury i naprężeń oleju.
Ten ostatni przyczynia się do niebezpieczeństwa lokalnych formacji węgla. Dlatego w lotnictwie wojskowym w przyszłości należy zrezygnować ze stosowania olejów na bazie estrów. W tym celu najbardziej obiecujące są oleje nowego typu – oparte na prostych perfluoroalkilopolieterach. Według współczesnych danych związki te są nietoksyczne i są nawet stosowane za granicą w perfumerii oraz do konserwacji marmurowych zabytków sztuki i architektury.
Dodatki mają duży wpływ na właściwości środowiskowe smarów. W olejach lotniczych szeroko stosowane są jako dodatki takie tradycyjne antyoksydanty i inhibitory korozji, jak dioktylodifenyloamina, fenylo-p-naftyloamina, benzotriazol, dodatek sukcynoimidowy K-51 i inne.
Na całym świecie od dawna trwają prace nad stworzeniem nowych, nietoksycznych i biodegradowalnych produktów. W szczególności od lat 90. opracowano substytuty dodatków zawierających chlor. Ważną kwestią jest wymiana związków ołowiu. Zamiennikami ołowiu są związki bizmutu. Rozpoczęto opracowywanie dodatku bizmutoditiokarbaminianowego.
Dodatki takie jak Mif-1 (dodatek typu benzenu o złożonym składzie), Irganox L-57 (dodatek przeciwutleniający z Siba, oktylowana i butylowana difenyloamina), dodatek X (związek zawierający fluor z grupami funkcyjnymi tlenosiarczynowymi i hydroksykarbaminianowymi) i inne został opracowany.
Poprawiane są właściwości znanych dodatków. Tak więc w fosforanie trikrezylu zawartość neutrotoksycznego ortoizomeru jest zmniejszona do 3% (Rosja), aw USA wytwarzany jest fosforan trikrezylu, który nie zawiera ortoizomeru.
Zagrożenie pożarowe i wybuchowe paliw anapaliwnych i smarów
Obecnie stosowane paliwa lotnicze i smary są produktami palnymi. W warunkach pożarowych szczególnie niebezpieczne są paliwa gazowe. Paliwa węglowodorowe (paliwa do silników odrzutowych, benzyny itp.) to ciecze palne (ciecze palne). Charakteryzują się wysoką produkcją ciepła (-2000 ° C) i lotnością, łatwo tworzą mieszanki palne z powietrzem, które po spaleniu tworzą dużą ilość produktów spalania (duży współczynnik stechiometryczny), które są dobrymi dielektrykami, a co za tym idzie, może gromadzić ładunki elektryczności statycznej.
Ze względu na zagrożenie pożarowe ciecze palne dzielą się na trzy kategorie. Temperatura zapłonu służy jako wskaźnik określający (określa się ją zgodnie z GOST 12.1.044-89):
W zależności od temperatury samozapłonu (określonej zgodnie z GOST 12.1.044-89) paliwa węglowodorowe należą do jednej lub drugiej grupy wybuchowej mieszaniny oparów z powietrzem:
Odważamy się, że opary paliw węglowodorowych z powietrzem należą do kategorii wybuchowej TTA: określa się ją zgodnie z GOST 12.1.011-78. Wskaźnik ten wykorzystywany jest przy wyborze rodzaju przeciwwybuchowego sprzętu elektrycznego oraz przy projektowaniu gaśnic.
O właściwościach palnych paliwa decydują również granice stężenia zapłonu (CIL) - minimalna i maksymalna zawartość oparów paliwa w mieszaninie z powietrzem (utleniaczem), przy której możliwe jest rozprzestrzenianie się płomienia przez mieszaninę w dowolnej odległości od zapłonu źródło (GOST 12.1.044-89). Ważną cechą paliwa są granice temperatury zapłonu - temperatury, w których pary nasyconego paliwa w powietrzu mają stężenia równe odpowiednio dolnemu lub górnemu CPV. Ogromne znaczenie ma minimalna energia wyładowania elektrycznego wymagana do zapalenia mieszanki para-powietrze.
Przy ocenie zagrożenia pożarowego przy obchodzeniu się z paliwami określa się również wskaźnik wypalenia – ilość paliwa spalanego w jednostce czasu z powierzchni jednostki; minimalna energia zapłonu - aby zapewnić bezpieczeństwo iskier elektrostatycznych. Ocenia się oddziaływanie palącego się paliwa z wodno-pianowymi środkami gaśniczymi (zgodnie z GOST 12.1.044-89).
Pożar często poprzedzony jest wybuchem mieszanki gazowo-powietrznej. Wraz z wybuchem mieszanin powietrza w rurach o dużej średnicy i długości może nastąpić spalanie detonacyjne, propagujące się z prędkością 1100-1400 m/s. W takim przypadku ciśnienie może wzrosnąć do 0,8 MPa lub więcej. Fala uderzeniowa o dużej prędkości powoduje gwałtowny wzrost ciśnienia, temperatury i gęstości mieszanki palnej, co z kolei przyspiesza chemiczne reakcje spalania i potęguje efekt destrukcyjny.
Wybuchowe stężenia par paliwa z powietrzem mogą tworzyć się w szerokim zakresie temperatur, a zwłaszcza w zamkniętych przestrzeniach i pojemnikach. Charakter i treść środków ostrożności regulują specjalne instrukcje resortowe. Istotą środków ostrożności jest zapobieganie powstawaniu źródła nagrzewania w miejscach powstawania mieszanin wybuchowych, a zwłaszcza źródła otwartego ognia. Jednym z najniebezpieczniejszych źródeł otwartego ognia jest wyładowanie potencjałów elektrostatycznych przez medium para-powietrze i powstawanie iskry po uderzeniu przez ciała stałe. Występowanie wysokich potencjałów elektrycznych w paliwie tłumaczy się jego właściwościami elektrofizycznymi. Charakteryzują się zdolnością kumulowania ładunków w objętości (podatność na elektrolizę) i ładują właściwości relaksacyjne (przewód elektryczny do nich).
W tabeli. 1.5. podano wskaźniki charakteryzujące właściwości pożarowe paliw lotniczych.
Tabela 1.5
Właściwości palne paliw lotniczych
1 Obliczone przez addytywność.
^ Obliczono zgodnie z równaniami (47) i (48) GOST 12.1.044-89 w oparciu o początkową temperaturę wrzenia -10/-4°C.
° W liczniku - w tyglu zamkniętym, w mianowniku - w tyglu otwartym. „Granice propagacji płomienia zgodnie z GOST 10277-89.
Normalna prędkość płomienia
Szybkość rozprzestrzeniania się płomienia w mieszaninie palnej zależy od warunków jego wyznaczania i liczenia. Do oceny porównawczej paliw według tej charakterystyki przyjęto normalną prędkość rozchodzenia się płomienia – jest to liniowa prędkość ruchu strefy spalania względem świeżej jednorodnej mieszanki palnej w kierunku prostopadłym do czoła płomienia. Szybkość rozprzestrzeniania się płomienia w takich warunkach dla danego składu mieszaniny palnej można uznać za charakterystykę fizykochemiczną zależną jedynie od ciśnienia i temperatury.
Eksperymentalnie normalną prędkość propagacji płomienia określa się zgodnie z GOST 12.1.044-89.
W temperaturze 20 ° C i ciśnieniu 0,101 MPa w mieszaninach węglowodór-wodór-powietrze maksymalna prędkość u osiągana jest przy stężeniu paliwa w mieszaninie C ^ ~ 1,15 C st x (ryc. 1.24), tj.
z a - 0,87 i liczbą atomów węgla w węglowodorze n\u003e 7 wynosi -39-40 cm / s (ryc. 1.25). Minimalna normalna prędkość rozchodzenia się płomienia i masowa prędkość spalania osiągane w granicach stężeń rozchodzenia się płomienia w normalnych warunkach wynoszą odpowiednio 4-6 cm/s i (5-7) 10° g/(cm 2 s).
W przypadku braku danych doświadczalnych normalną prędkość propagacji płomienia należy dobrać przez interpolację z wartości u dla mieszanin o podobnych właściwościach fizykochemicznych lub zastosować równania empiryczne. Proste i wygodne równania zaproponował A.S. Jazda przed jazdą:
- (1.3)
t \u003d t p + B (St-C ^ (C w -C t),
gdzie u jest prędkością propagacji w cm/s; m jest masową szybkością spalania mieszaniny, g/(cm2s); i 11P, tn - graniczne (minimalne) wartości prędkości propagacji płomienia; С i С n to stężenie paliwa w mieszaninie w dolnej i górnej granicy stężeń rozprzestrzeniania się płomienia; A i B to współczynniki wyznaczone z jednego punktu doświadczalnego.

Ryż. 1.24.
propagacja płomienia w zależności od molowego współczynnika stechiometrycznego nadmiaru powietrza bm:
- - parafina; * - olefinowy; ° - acetylen; D - olej; © - dipolarny; ° węglowodory o C p 11 2 „cykle”
- 1 2 3 4 5 b 7 p

Ryż. 1,25. Maksymalna normalna prędkość rozprzestrzeniania się płomienia w mieszaninie paliwowo-powietrznej w zależności od liczby atomów węgla w cząsteczce węglowodoru (P=0,101 MPa, 1=20°C, otwarta szklana rurka: długość 57 cm, średnica 2,5 cm): - parafina; * - olefinowy;
° - acetylen; D - naftenowy; w - dnolsfipovye; o cykliczny (C P P2 ");
1 - benzyna [ 116]; 2 - benzen
Zależność funkcjonalną między prędkością propagacji płomienia a stężeniem paliwa C t przy C t C * t (ale daną przez EMIN) można przedstawić równaniem:
- - = 11 p
/ s r -s; ja
"s t - s "t"
gdzie m i oraz n- normalna prędkość propagacji płomienia
przy stężeniach paliw w mieszaninie C t i C*t, cm/s; i pp- też,
przy dolnej granicy stężenia rozchodzenia się płomienia, cm/s.
Przybliżony przebieg krzywej oraz n - /(C t) w mieszaninie złożonej
skład można budować na trzech punktach odniesienia odpowiadających dolnej i górnej granicy stężeń oraz maksymalnej prędkości rozprzestrzeniania się płomienia. Dla tych punktów muszą być znane stężenia paliwa i prędkości rozprzestrzeniania się płomienia.
Wartości C t i i i za określone punkty są obliczane
w następujący sposób. Każda złożona mieszanina gazów palnych jest reprezentowana jako składająca się z odpowiedniej liczby prostych mieszanin. Obliczenie składu w granicach stężeń i w punkcie maksymalnych prędkości odbywa się zgodnie z zasadą mieszania, w oparciu o granice stężeń i skład „mieszanek maksymalnych”. Odpowiednie równanie obliczeniowe ma postać:
C] + C* 2 + Su uh...
- -Ja---g...
- (1.5)
gdzie b- stężenie paliwa na CPRP lub w mieszaninie o maksymalnej prędkości rozprzestrzeniania się płomienia,% (obj.); C, C 2, C 3, ... - stężenie prostych gazów w złożonej mieszaninie,
(s, + C2 + C3 + ... = 100%); b|, b 2 , b 3> ... - stężenie gazów w mieszaninach prostych na KPRP lub w mieszaninach z oraz oraz % (obj.).
Wartość maksymalnej normalnej prędkości rozprzestrzeniania się płomienia w mieszaninie oblicza się z równania;
C, r/, + C2u2 + C3u3 +
C, + C 2 + C 3 4-...
- (1.6)
gdzie C*, C 2 , C 3 - zawartość prostych mieszanin w złożonej mieszaninie z maksymalną prędkością propagacji płomienia,% (obj.); oraz*, i 2 , a 3 to maksymalne prędkości propagacji płomienia w prostych mieszaninach, cm/s.
Aby obliczyć inne punkty krzywej i i= /(C; .) należy ustawić kilka dowolnych wartości prędkości płomienia, znaleźć stężenie b w złożonej mieszaninie zgodnie z równaniem (1.5), w którym C, C 2 , C 3 są podane przez skład mikstura.
Ta metoda obliczeniowa ma zastosowanie do mieszanin gazowych o podobnym charakterze (np. metan-propan). Ta technika nie ma zastosowania do mieszaniny S P N W z H3 i CO.
Masowe tempo spalania jest wprost proporcjonalne do bezwzględnej temperatury podgrzewania mieszanki i można je obliczyć z równania:

gdzie w, wtedy i t „Reo”- masowe tempo spalania mieszanki w temperaturze T, To i T P r e d odpowiednio g/(cm-s).
Jeśli T»T pre e D, to

Zależność maksymalnej normalnej prędkości propagacji płomienia od temperatury i ciśnienia w przybliżeniu opisuje równanie:
oraz' =u1(T/273) 2 ?(/’/10 5)", (19)
gdzie u'o to maksymalna normalna prędkość rozchodzenia się płomienia w temperaturze 293 K i ciśnieniu 0,101 MPa, cm/s; T jest temperaturą l płomienia w K; P - ciśnienie, w Pa; p - wykładnik, ns w zależności od ciśnienia w zakresie MO 4 + 5-10 5 Pa; dla mieszanki paliwowo-powietrznej n = -0,3 -*? -0,4; dla mieszanin węglowodór-tlen P = -0,1 -5-0.
Maksymalna normalna prędkość propagacji płomienia w funkcji stężenia tlenu w utleniaczu P R P Wu P
giil = \%ig" 0 + B-
gdzie G „Ja! Ale - w y, p r^ 0 , cm2/s; B - współczynnik określony na podstawie danych eksperymentalnych (dla propanu B ~ 0,22); u/ t- wyjątkowo niskie stężenie tlenu w utleniaczu.
Wartość u*n przy różnych stężeniach tlenu w utleniaczu 1 //"P gdy temperatura podgrzewania mieszaniny zmienia się od 310 do 422 K, można ją wyznaczyć za pomocą równania:
":=w; (u,-s), (MO
gdzie u*n - w cm/s; T - w K; А, С ip - znajdują się na podstawie danych eksperymentalnych, ich wartości dla propanu, izooktanu i etylenu podano poniżej:
Granice stężenia i temperatury rozprzestrzeniania się płomienia
Granice stężeń rozprzestrzeniania się płomienia (KPRP) w mieszaninie palnej to graniczne minimalne i maksymalne stężenia paliwa w mieszaninie, przy których rozprzestrzenianie się płomienia jest nadal możliwe (odpowiednio dolna i górna granica). Zależą one od aktywności chemicznej paliwa, stężenia utleniacza i zanieczyszczeń obojętnych, przewodności cieplnej i pojemności cieplnej mieszanki, temperatury i ciśnienia. KPPR dla paliw zawiesinowych, w oparciu o ich właściwości fizykochemiczne, są determinowane przez medium dyspersyjne. Oznaczanie KPRP dla jednorodnych mieszanin palnych przeprowadza się zgodnie z GOST 12.1.044-89: zgodnie z punktem 4.11 eksperymentalnie i zgodnie z punktem 4.12 - metodą obliczeniową.
Zgodnie z GOST 12.1.044-84 granice stężenia propagacji płomienia są określone jako

gdzie Cn (i) - niższy (górny) CPRP,% (obj.); R- współczynnik stechiometryczny (liczba moli tlenu na mol paliwa); a oraz b- stałe uniwersalne, ich wartości podano poniżej:
Dla paliw C P N W
P \u003d n + t / 4.
Błąd obliczeń: dla dolnej granicy 0,12; dla górnych 0,40 at (3 p > 7,5. Dane w KIRP w zależności od R(% obj.) podano w tabeli. 1,6 (GOST 12.1.044-84).
Tabela 1.6
Granice stężeń rozprzestrzeniania się płomienia (dolny i górny) par i gazów w powietrzu
Istnieją inne równania do obliczania CPRP, a mianowicie:
- 4,76-(N-1) + ! '
- (1.14)
- 4,76/Y +4'
- (1.15)
gdzie С„ i Od do - w około.); N to liczba atomów tlenu wymagana do całkowitego utlenienia paliwa.
Na paliwo С„Н t
- (1.17)
- 3,74 10 5
gdzie Cn - w% (obj.); () n to najniższa molowa wartość opałowa, kJ/kmol.
Dla paliw węglowodorowych SpN t przy 3 p 10 błąd obliczeniowy wynosi ±15%.
Jeżeli znane są KRI dla poszczególnych składników paliwa, to zaleca się obliczenie jego niższego KRI za pomocą równania:
gdzie C i Cn są stężeniami pierwszego składnika w mieszaninie i przy dolnej granicy, % (obj.).
Dla paliw C p N t w pierwszym przybliżeniu a k ~ a p - 1.42. Przeliczenie i od w do jakiś oraz jakiś wytworzony:

gdzie Cn (d) to stężenie paliwa w dolnej (górnej)
KPRP, % (obj.); Mt i Mo to masa cząsteczkowa paliwa i utleniacza; Bo - w kg utleniacza/kg paliwa; bm to molowy współczynnik stechiometryczny, mol paliwa/mol paliwa.
Ponowne obliczenie dolnego KPPR dla różnych temperatur można przeprowadzić według równania:
L II I
T - 293
gdzie Tn jest temperaturą (w K) produktów spalania mieszanki, w której stężenie paliwa przy 293 K odpowiada niższemu KPP (w pierwszym przybliżeniu Tn dla mieszanki węglowodorowo-powietrznej wynosi 1600-1650 K); C „ i C „ - stężenia paliw odpowiadające dolnej granicy stężeń w temperaturach T i 293 K, % (o.).
Równanie (1.20) jest ważne w szerokim zakresie temperatur, ale nie może być stosowane w temperaturach zbliżonych do temperatury samozapłonu.
Temperaturę produktów spalania przy niższym KPRP można również obliczyć za pomocą równania
- (A. + 1) -s_s
- (1.21)
Steha
gdzie Tn w K; T z temperaturą mieszaniny przed spalaniem, K; Сstsh - stężenie paliwa w mieszaninie o składzie stechiometrycznym, % (obj.);
Срш to średnia izobaryczna pojemność cieplna produktów spalania w temperaturze T, „kJ / (kg ° С).
CRP jest praktycznie niezależny od wymiarów cylindrycznego naczynia reakcyjnego, jeśli jego średnica jest większa niż 50 mm, a kulistego, jeśli objętość przekracza 2000 cm3.
W celu określenia KPPR i optymalnego składu mieszaniny węglowodorowo-powietrznej wykresy pokazane na ryc. 1.26.
С„,s,%(ov.)

Ryż. 1.26. Granice stężeń rozchodzenia się płomienia w mieszaninach węglowodor-powietrze (Cb i C") oraz stężenia węglowodorów w mieszaninach o składzie stechiometrycznym (Cc,") w zależności od molowego współczynnika stechiometrycznego 1^m przy H20 ° C P = 0,101 MPa:
- - parafina; a - olefinowy;
- ? - naftenowy; ? - aromatyczny
Palne mieszanki par paliwa z powietrzem w przestrzeni nad paliwem mogą powstawać tylko w określonym zakresie temperatur. Minimalna temperatura, w której palna mieszanina zdolna do stacjonarnego spalania po zapaleniu ze źródła zewnętrznego może nadal tworzyć się w zamkniętej objętości przestrzeni nadpaliwowej, nazywana jest dolną granicą temperatury; odpowiada to niższemu KPP. Najwyższa temperatura, w której mieszanina oparów z powietrzem w przestrzeni nad paliwem nadal zachowuje zdolność do spalania stacjonarnego, nazywana jest górną granicą temperatury; odpowiada to górnemu KPRP Eksperymentalne określenie granic temperatury tworzenia mieszanin wybuchowych przeprowadza się zgodnie z GOST 12.1.044-89 (s. 4.12), obliczone - zgodnie z zastosowaniem tej samej normy.
Temperatura, w której osiągana jest dolna granica temperatury tworzenia mieszaniny wybuchowej pod ciśnieniem atmosferycznym, jest zwykle identyfikowana z temperaturą zapłonu. W punkcie zapłonu pali się tylko powstała mieszanina para-powietrze, ale proces spalania nie stabilizuje się.
Obliczenie granicznych temperatur dla tworzenia mieszanin palnych sprowadza się do następujących operacji. Początkowo przy danym ciśnieniu całkowitym P i znanych wartościach współczynnika nadmiaru utleniacza (powietrza) odpowiadającego dolnemu i górnemu KPRP (a n i c), zgodnie z równaniem (1.22) wyznacz
ciśnienia cząstkowe par paliwa Р t :
X | 0,232 o? 0 mln t " ?« -
gdzie P jest ciśnieniem całkowitym, Pa; C - współczynnik stechiometryczny, kg utleniacza/kg paliwa; a - współczynnik nadmiaru utleniacza; Mt to masa mola paliwa, kg/kmol; Mo to masa mola środka utleniającego, dla powietrza Mo = 28,966 kg / kmol; w/ 0 - stężenie tlenu w utleniaczu według masy.

Ryż. 1.27.
Następnie zgodnie z tabelami lub wykresami Pc.p = ^ (0 (gdzie P, ciśnienie par paliwa nasyconego) znajdź temperatury odpowiadające obliczonym wartościom Pt-
Jeżeli granice stężeń dla tworzenia mieszanin palnych są nieznane, granice temperatury można w przybliżeniu obliczyć za pomocą równania:
1,15 1*(7,5 R d) - 0,239 3,31
gdzie ja - w 0 C; 15% - temperatura wrzenia frakcji 5%, 0 C; Рт - ciśnienie par paliwa w KPP (Р lub Р), kPa; 8 „ z „ - entropia parowania w temperaturze 15% i ciśnieniu atmosferycznym (przyjęta zgodnie z wykresem na rys. 1.28).

Ryż. 1.28.
60 80 100 120 140 160 180 1,°С
Energia palna i granice stężenia palnego
Palność jednorodnej mieszaniny palnej przez zewnętrzne źródło ciepła charakteryzuje się granicami stężenia i energią potrzebną do jej zapalenia.
Stężenia graniczne zapłonu (CFL) to takie graniczne stężenia paliwa w mieszance, przy których lokalne źródło zapłonu (wyładowanie elektryczne, nagrzany korpus, płomień) jest w stanie zapewnić rozprzestrzenienie się procesu spalania na całą objętość mieszanki. Analogicznie do KG1RP rozróżnia się dolny i górny CPV. Zależą one od właściwości fizykochemicznych paliwa i utleniacza, energii i rodzaju źródła zapłonu, jego lokalizacji itp.
Według Ya.B. Zeldovicha, energia potrzebna do zapalenia jednorodnej mieszaniny palnej jest określona przez:
R1-T z r (T 2 -T c)
gdzie pc i T c są gęstością i temperaturą mieszaniny; T g jest temperaturą produktów spalania w początkowej komorze spalania; L 7 - współczynnik przewodności cieplnej produktów spalania przy Tg; u - normalna prędkość propagacji płomienia; C rt - średni
izobaryczna masowa pojemność cieplna gazu w kulistej warstwie 8 T otaczającej kulistą komorę spalania początkowego; 5, - szerokość termiczna czoła płomienia.
Równanie (1.24) ma również zastosowanie w przypadku zapłonu poruszającej się mieszaniny, jeżeli współczynnik przewodności cieplnej L 7 zostać zastąpione przez turbulentny współczynnik wymiany IV/"(/ - skala
turbulencja, W/*- prędkość pulsacyjna), a wartość n - prędkość propagacji płomienia w przepływie turbulentnym.
Skład mieszaniny odpowiadający minimum krzywej O = KS,), nazywa się optymalnym. Dla normalnych węglowodorów parafinowych stężenie paliwa w mieszaninie o optymalnym składzie w temperaturze 25°C można wyznaczyć ze stosunku:
- 1 - metan; 2 - etan; 3 - propan;
- 4 - n-butan; 5 - n-heksan; 6 - n-heptan;
- 7 - cyklopropan: 8 - eter dietylowy;
- 9 - benzen
Wraz ze wzrostem stężenia tlenu w utleniaczu, optymalny skład mieszanki palnej przesuwa się w rejon niższego stężenia paliwa.
Zależność optymalnej (minimalnej) energii zapłonu od ciśnienia i temperatury mieszanki palnej opisuje równanie [114]:
O-opt


gdzie Oopt jest energią zapłonu w P i T, J; Cb - energia zapłonu przy T = 273 K i P = 10 5 Pa.
Równanie (1.26) ma dobrą korelację z danymi eksperymentalnymi.
Zależność optymalnej energii zapłonu od stężenia tlenu w utleniaczu opisuje równanie

gdzie (С? 0 „„,) y / = / - optymalna wartość energii zapłonu mieszanki paliwowo-tlenowej; ~ stężenie objętościowe
tlen w utleniaczu; n jest wykładnikiem, jest bliski jedności (n ~ 0,8).
Dane eksperymentalne dla metanu, etanu i propanu przy wymianie c/ x, od 0,1 do 0,21 i ciśnienia od 0,98 do 19,6 kPa potwierdzają równanie (1,27). Najwyraźniej zachowuje ważność dla mieszanin węglowodorów.
Stężenia paliwa na granicach zapłonu można obliczyć, jeśli KPRP i wartości () ref i C opt są znane zgodnie z równaniami

o.5 (s; + s;) \u003d C_ + 0,15 (C. (1,29)
Równania (1.28) i (1.29) obowiązują dla --
Oznaczając właściwe części tych równań, odpowiednio B i 0,5A, otrzymujemy
Z" - Z" = B i C"+ C” = ALE . (1.30)
C” = 0,5(L-B) i C; =0,5 (A + B). (1.31)
W powyższych równaniach: C in i C n - stężenia paliw w mieszaninie przy górnym i dolnym KPRP; C in i C” - stężenie paliwa w mieszance przy górnym i dolnym CPV z energią zapłonu pojemnościowego ładunku elektrycznego; C opt - stężenie paliwa w mieszance odpowiadające O ref.
Równania (1.28) i (1.29) oparte są na wynikach badań eksperymentalnych przedstawionych na ryc. 1.30.
- (s;-s>;)-2s opt

Ryż. 1.30. Powierzchnia zapłonu mieszanin C p N P1 + 02 + ^ w zależności od energii zapłonu
Granice stężeń zapłonu zależą od natężenia przepływu, zbliżając się do siebie wraz z jego wzrostem (ryc. 1.31 i 1.32).
Wpływ prędkości przepływu na energię zapłonu poprawnie opisuje równanie:
(2 = (?o + Au "do (1.32)
gdzie (Zo - energia zapłonu mieszaniny stacjonarnej, 10 "3 J; XV - prędkość przepływu, m / s; A - współczynnik ustalony eksperymentalnie.

Ryż. 1.31.

Ryż. 1.32. Współczynnik nadmiaru powietrza a przy CPV mieszanki benzyna-powietrze w zależności od natężenia przepływu? i ciśnienie Р [ 114]:
Temperatura zapłonu i temperatura samozapłonu
Temperatura zapłonu to minimalna temperatura, w której powstała mieszanina para-powietrze może ulec zapłonowi przez zewnętrzne źródło ciepła, ale proces spalania nie jest ustabilizowany. Eksperymentalnie temperaturę zapłonu określa się w otwartym lub zamkniętym tyglu zgodnie z GOST 12.1.044-84 (pozycje 4.3 i 4.4). Obliczone określenie temperatury zapłonu odbywa się zgodnie z GOST 12.1.044.84 (punkt 4.5).
Temperatura zapłonu jest o 10-15°C niższa od temperatury granicznej, w której powstaje palna mieszanina zdolna do rozprzestrzeniania się płomienia.
Do przybliżonego określenia temperatury zapłonu można wykorzystać zależność pokazaną na rys. 1. 1.33.

Ryż. 1.33. Temperatura zapłonu 1 V cp paliw do silników odrzutowych i benzyny B-70 w zależności od prężności par nasyconych Pn p przy 1=40°C w tyglu zamkniętym (62]): o - paliwa o różnym składzie - krzywa uogólniająca
Samozapłon to proces zapłonu mieszanki palnej bez kontaktu z płomieniem lub gorącym ciałem. Minimalna temperatura początkowa wystarczająca do samozapłonu mieszanki palnej nazywana jest temperaturą samozapłonu. Zależy to od chemicznego charakteru paliwa, składu mieszanki paliwowo-powietrznej, ciśnienia, adiabatycznego charakteru procesu samozapłonu, obecności katalizatorów i inhibitorów utleniania oraz innych czynników.
Odstęp czasu pomiędzy osiągnięciem przez mieszankę palną temperatury samozapłonu a pojawieniem się płomienia nazywany jest okresem opóźnienia samozapłonu. Dostarczając paliwo płynne obejmuje proces atomizacji, nagrzewania i parowania kropel paliwa, dyfuzji oparów paliwa i tlenu, a na końcu reakcji chemicznych.
Temperatura i czas opóźnienia samozapłonu są powiązane zależnością:

gdzie mi- efektywna energia aktywacji, kJ/kmol; mi\u003d 8,31419 kJ / (kmol K) - uniwersalna stała gazowa; t- okres opóźnienia samozapłonu w temperaturze T.
Skłonność węglowodorów i ich mieszanin do samozapłonu charakteryzuje się minimalną temperaturą samozapłonu uzyskaną w warunkach adiabatycznych, gdy czas ekspozycji mieszaniny palnej w danych warunkach początkowych nie ogranicza procesu samozapłonu.
Minimalna temperatura samozapłonu jest jednoznacznie określona przez strukturę cząsteczki. Tak więc, na przykład, dla węglowodorów parafinowych, pierwsza jest bezpośrednio związana z efektywną długością łańcucha węglowego Lc, którą oblicza się za pomocą równania:
- 21>GLG,
- (1.34)
gdzie r jest liczbą grup CH3 w cząsteczce; k to liczba łańcuchów węglowych rozpoczynających się i kończących na grupie CH3, m* to liczba możliwych łańcuchów zawierających b^-atomów węgla. Zależność 1 sv =A(bc) pokazano na ryc. 1.34.

Ryż. 1.34.
- 1 - CH 4; 2-C2H6; 3 - C3H ”; 10 - n - C4H10; 11-n-C5H12;
- 14 - n - SL N M; 15 - n - C7H16; 16 - n - SkNsch; 17 - n - SdN 2o;
- 18 - n - C| 0H22; 19-n-C,2H2I; 21 - n - C14H30; 22 - n - C|^H 3 4
Temperatura samozapłonu mieszanin węglowodorów nie jest zgodna z zasadą addytywności, zwykle jest niższa niż obliczona na podstawie określonej reguły.
Dane dotyczące temperatury samozapłonu mieszanin powietrzno-paliwowych o optymalnym składzie w zależności od liczby atomów węgla w cząsteczce węglowodoru (dla paliw odrzutowych w powyższym wzorze) przedstawiono na rys. 2. 1.35. Wpływ ciśnienia i stężenia tlenu w utleniaczu ilustrują dane pokazane na ryc. 1.36.

Ryż. 1.35. Zależność temperatury samozapłonu mieszanin powietrzno-paliwowych o optymalnym składzie od liczby atomów węglowodorów n w cząsteczce przy Р=0,101 MPa [124]; t jest okresem opóźnienia samozapłonu; t L - "o; R.T. - paliwa do silników odrzutowych (p-w powyższym wzorze) - parafina; a-olefinowy; ? - węglowodory naftenowe

Ryż. 1.36. Zależność temperatury samozapłonu paliwa T-6 od ciśnienia P i stężenia tlenu w utleniaczu f 0 2 (wg W. W. Małyszewa):
2 = 0 2/(°2+L, d)
Temperatura samozapłonu zależy od zdolności paliwa do tworzenia palnych mieszanin w fazie gazowej. Wynika z tego, że temperatura samozapłonu zawiesiny
paliwa zależy od ośrodka dyspersyjnego i zagęszczacza. Faza rozproszona bierze udział w procesie samozapłonu tylko w zakresie pochłaniania ciepła, gdy zawiesina jest ogrzewana do temperatury samozapłonu fazy ciekłej.
Ciśnienie wybuchu w zamkniętej objętości
Ciśnienie wybuchu - najwyższe ciśnienie, jakie występuje podczas wybuchu deflagracji mieszaniny para-powietrze w zamkniętej objętości przy początkowym ciśnieniu 0,101 MPa. Szybkość narastania ciśnienia podczas wybuchu jest pochodną ciśnienia wybuchu w funkcji czasu (s1R/(1t) na wstępującym odcinku zależności P=Y t).
Eksperymentalnie maksymalne ciśnienie wybuchu i szybkość wzrostu ciśnienia podczas wybuchu mieszanin parowo-powietrznych określa się zgodnie z GOST 12.1.044-89 (Załącznik 8). Obliczone określenie szybkości wzrostu ciśnienia podczas wybuchu przeprowadza się zgodnie z GOST 12.1.044-89 (dodatek 12).
Ciśnienie wybuchu jest określane przez:

gdzie Pvzr - ciśnienie wybuchu, Pa; Pn - ciśnienie początkowe, Pa; T” i T p.s. - temperatura początkowa i temperatura produktów spalania. DO; kolec - liczba moli produktów spalania i początkowa mieszanina.
Maksymalna szybkość wzrostu ciśnienia (w Pa/s) jest obliczana z równania

gdzie Ro jest ciśnieniem początkowym. Rocznie; u - normalna prędkość propagacji płomienia przy Po i To m/s; To jest początkową temperaturą mieszaniny, K; r jest promieniem bomby, m; P - R m /P 0 - obniżone maksymalne ciśnienie wybuchu; k jest wskaźnikiem adiabatycznym dla badanej mieszaniny; mi- wskaźnik termokinetyczny w zależności od i n, ciśnienia i temperatury; jeśli wartość mi nieznany, przyjmuje się, że jest równy 0,4.
Średnie tempo wzrostu ciśnienia (w Pa/s) oblicza się ze wzoru:
„s1R _ ZR 0 oraz ‘(i-)-i k * e ^m) z g / (l, k, e)
gdzie ^m,k 7 mi)-funkcja, jej wartość znajduje się zgodnie z nomogramem rys. 1.37.

Ryż. 1.37. Zależność funkcji /(n, u.w.) z obniżonego ciśnienia n=P/Pk,„ wykładnik adiabatyczny do i indeks termokinetyczny z mieszanina testowa (załącznik do GOST 12.1.044-84)
Wartości tg a k znajduje się za pomocą obliczeń termodynamicznych lub. w przypadku niemożności obliczenia, zaakceptuj do= 9,0 i k=1,4.
Nagłe i nagłe wypadki
Wypadek to niebezpieczne zdarzenie wywołane przez człowieka, które stwarza zagrożenie życia i zdrowia ludzi na obiekcie, określonym terytorium lub obszarze wodnym i prowadzi do zniszczenia budynków, konstrukcji, wyposażenia i pojazdów, zakłócenia procesu produkcyjnego lub transportowego , a także szkody w środowisku naturalnym (GOST R 22.0 .05-94).
Wypadek to destrukcyjne niekontrolowane uwolnienie energii lub składników aktywnych chemicznie (biologicznie, radiacyjnie). W zależności od źródła występowania rozróżnia się sytuacje awaryjne (ES) o charakterze naturalnym, sztucznym i naturalnym. Na ryc. 1,38 pokazuje względny wzrost liczby wypadków i katastrof naturalnych, spowodowanych przez człowieka i naturalnych spowodowanych przez człowieka w Rosji. Na ryc. 1,39 pokazuje dynamikę liczby wszystkich wypadków spowodowanych przez człowieka w Rosji w latach 1990-94. Z wykresu widać, że wzrost liczby sytuacji kryzysowych nie następuje płynnie, ale gwałtownie, z wybuchami pojawiającymi się w okresach bezpośrednio po wstrząsach społecznych (sierpień 1991, październik 1993).
W ostatnich latach szczególnie gwałtownie wzrosła liczba wypadków spowodowanych przez człowieka, w tym w lotnictwie.
Potencjalnymi obiektami wypadków są statki powietrzne, a także znajdujące się na terenie lotniska magazyny i magazyny wybuchowych i łatwopalnych produktów naftowych, punkty tankowania i obsługi technicznej oraz punkty napraw. Przyczyną sytuacji awaryjnych mogą być wycieki oleju.
produkty poprzez zespoły uszczelniające zaworów odcinających, pomp transferowych, rurociągów i urządzeń napełniających; poprzez wentylację przestrzeni gazowej zbiorników; przelewanie się zbiorników, cystern i zbiorników; czyszczenie zbiorników; zniszczenie korozyjne zbiorników i komunikacji.
Do przechowywania i transportu produktów naftowych wykorzystywane są różne pojemniki. O bezpiecznej eksploatacji kontenerów decyduje ich wytrzymałość. Jednak wypadki na tego typu obiektach mogą wystąpić z powodu niedociągnięć w istniejącym systemie monitorowania i monitorowania stanu obiektów, a także braku dokumentacji regulacyjnej i technicznej.
Bezpieczeństwo eksploatacji obiektów magazynowych produktów naftowych musi być zapewnione na etapie projektowania, budowy i eksploatacji. Takie podejście jest podyktowane analizą dokumentacji odbiorowej i eksploatacyjnej, a także przyczyn sytuacji awaryjnych. Ważnym zadaniem, którego rozwiązanie wpłynie na poprawę niezawodności eksploatowanych obiektów magazynowych, jest przeprowadzenie ich naukowo uzasadnionych kompleksowych badań technicznych oraz wyposażenie ich w system diagnostyki i monitoringu eksploatacyjnego stanu konstrukcji metalowych, fundamentowych, termoizolacyjnych oraz sprzęt procesowy.
Dla bezpiecznego zarządzania przepływami produktów ropopochodnych duże znaczenie ma sprawność armatury technologicznej rurociągu: odcinająca, dławiąca, urządzenia zabezpieczające; Zawory regulacyjne; zawory odwrotnego działania (aby zapobiec możliwości przemieszczania się produktu, odwrotność pracownika); armatura awaryjna i odcinająca (do automatycznego odcięcia dopływu do odcinka awaryjnego lub jego wyłączenia), spusty kondensatu itp.
Liczba wypadków

Ryż. 1.38.
- 1 - pg "krewni;
- 2 - naturalno-technologiczny;
- 3 - technogeniczny

Ryż. 1.39.
Gdy sprzęt jest rozhermetyzowany, produkt wypływa i szybko odparowuje, tworząc stężenie
wybuchowych i palnych mieszanin gazowo-parowo-powietrznych. Przypadkowe emisje lub wycieki mieszanin para-gaz prowadzą do tworzenia chmur, które mogą detonować. W pracy uwzględniono detonację układów parowo-gazowych i aerodyspersyjnych. Występowanie detonacji w dużych chmurach wyjaśniają następujące mechanizmy. Pierwsza z nich uwzględnia możliwy efekt intensywnego promieniowania cieplnego z długiego płomienia w chmurach wstępnie wymieszanych przez turbulentne przepływy gazu.
Drugi mechanizm powstawania detonacji polega na przyspieszaniu płomieni w dużych chmurach na skutek różnicy przyspieszeń objętości elementarnych spalonego gazu i świeżej mieszanki w płomieniu turbulentnym. Różnica ta powstaje pod działaniem średnich gradientów ciśnienia w płomieniu z powodu różnej wyporu elementarnych objętości gazu o różnych gęstościach, co prowadzi do dodatkowej turbulencji przepływu i sprzężenia zwrotnego. Ten mechanizm dodatniego sprzężenia zwrotnego, determinowany różnicą gęstości w różnych obszarach chmury, może znacznie zintensyfikować przyspieszenie płomienia.
Zapłonu towarzyszy jasny błysk w wysokiej temperaturze. Najbardziej akceptowalną figurą geometryczną rozdmuchanej mieszaniny para-gaz jest figura nieregularnej kuli lub elipsy (kula ognia). Przez kulę ognia (OS) rozumie się produkt nagłego odparowania lub wycieku zgazowanego paliwa (lub gazu), któremu towarzyszy ich rozbłysk, a następnie normalne lub deflagracyjne spalanie. Dla licznych wyładowań liniowych i cyklicznych palnych węglowodorów w zakresie gęstości od 700 do 1000 kg/m3 podano współczynniki dla średnicy kuli ognistej:
gdzie M to masa paliwa w OH, kg;
Тf - rzeczywista temperatura na OR (w chmurze), 0 С;
Trep - temperatura odniesienia (referencyjna), °С.
Zakres współczynnika 4,2n-5,3 zależy od rodzaju paliwa i warunków powstawania chmur.
Przez całe życie chmury podczas jej naturalnego spalania wyrażenie ma postać:
m = 0M-*1m-1±.
Zależności te pokazano na ryc. 1,40 i 1,41.

Ryż. 1.40.

Ryż. 1.41.
Istnieje duże niebezpieczeństwo wybuchu mieszanin para-gaz-powietrze w zamkniętej objętości. W tabeli. 1.7 pokazuje granice detonacji węglowodorów w powietrzu w zamkniętej objętości i otwartej przestrzeni, które wskazują na większe niebezpieczeństwo wybuchu mieszanin gazowych lub gazowo-parowych w zamkniętej objętości. Wyjaśnia to zarówno procesy przyspieszania reakcji spowodowane zwiększoną autokatalizą, jak i wzmacnianiem fal odbitych na początku procesu aryjskiego oraz szeregiem zawsze istniejących przyczyn kinetycznych. Zwiększona łatwość wzbudzania detonacji w naczyniach wynika ze zdolności ścianek do generowania turbulencji w przepływie przed płomieniem, co przyspiesza przejście od spalania do detonacji.
Granice detonacji węglowodorów w powietrzu
Wybuch nagromadzonej mieszanki gazowej może nastąpić pod działaniem przypadkowej iskry. Wybuch z powodu wyładowania statycznego jest również możliwy przy otwartym załadunku produktu naftowego, w szczególności w przypadku braku urządzenia uziemiającego. Najczęstszą przyczyną wybuchu jest iskra, w tym w wyniku nagromadzenia elektryczności statycznej. Iskra elektryczna może wystąpić bez żadnych przewodów i sieci. Jest niebezpieczna, ponieważ występuje w najbardziej nieoczekiwanych miejscach: na ścianach zbiorników, na oponach samochodowych, na ubraniach, podczas uderzenia, tarcia itp. Inną przyczyną wybuchu są zaniedbania i brak dyscypliny pracowników.
Tam, gdzie możliwe jest tworzenie się mieszanin para-gaz-powietrze, należy zapewnić niezawodną ochronę odgromową, ochronę przed elektrycznością statyczną oraz przewidzieć środki zapobiegające iskrzeniu urządzeń elektrycznych i innych urządzeń.
W przypadku wypadków związanych z wybuchami dochodzi do zniszczenia okolicznych obiektów i obrażeń ciała. Zniszczenie jest konsekwencją działania duchów produktów wybuchu i fali uderzeniowej powietrza. W tym przypadku głównymi czynnikami uszkadzającymi są fala uderzeniowa, promieniowanie cieplno-świetlne i ładunki toksyczne (tlenek węgla). Osoby znajdujące się w odległości 5 m doznają oparzeń I stopnia i innych obrażeń.
Wypadkom wybuchowym często towarzyszą pożary, które mogą spowodować katastrofalne skutki, a następnie większe eksplozje i większe zniszczenia. Przyczyny pożarów są zwykle takie same jak wybuchy. W takim przypadku wybuch może być przyczyną lub skutkiem pożaru i odwrotnie, pożar może być przyczyną lub skutkiem wybuchu.
Pożar to spontanicznie rozwijające się miasto renu, nie przewidziane przez procesy technologiczne. Spalanie produktów ropopochodnych może nastąpić w zbiornikach, sprzęcie produkcyjnym i wyciekach na otwartych przestrzeniach. W przypadku pożaru produktów naftowych w zbiornikach może dojść do wybuchów, zagotowania i ich uwolnienia, aw efekcie do rozlania gorącej cieczy. Emisje i zagotowanie produktów ropopochodnych są bardzo niebezpieczne, co wiąże się z obecnością w nich wody i charakteryzuje się szybkim spalaniem spienionej masy produktów. Podczas gotowania temperatura gwałtownie wzrasta (do 1500 ° C) i wysokość płomienia.
Do oceny stopnia uszkodzenia obiektu stosuje się zwykle tzw. krzywą progową, która odnosi strumień energii cieplnej i świetlnej q (strumień cieplny) oraz całkowitą energię O, przypadającą na jednostkę powierzchni (ryc. 1.42).

Ryż. 1.42.
Dla długich czasów narażenia termicznego, przekraczających czas ewentualnego istnienia obiektu w stanie nieuszkodzonym, próg uszkodzenia będzie określony wyłącznie przez strumień cieplny (światło cieplne) n. Przy impulsach o krótkiej ekspozycji próg będzie określany głównie przez energię O. Wartości R i O przekraczające próg spowodują bezwarunkowe uszkodzenie obiektu.
Jeśli wartość I lub O jest mniejsza niż ich wartości progowe, nie ma typowej zmiany i możliwy jest jedynie niewielki dyskomfort. Na przykład wraz ze wzrostem czasu trwania promieniowania z 0,5 do 2 s zmniejszam się ze 120 do 30 jednostek, tj. z niewielkim wzrostem O nawet przy 4-krotnym wzroście czasu ekspozycji, wpływającym na urazy
są nieobecne, a osoba może odczuwać tylko niewielki dyskomfort.
Jednak wartość całkowitej energii O przypadającej na obiekt zniszczenia w tym samym czasie wzrasta z około 10 do 25 jednostek. (^.
W ten sposób linia K, odpowiadając na powiązane zmiany w I i O, tworzy strefę (obszar) zmiany, wskazaną na rysunku po prawej stronie linii K.
Jedną z najbardziej nieprzyjemnych konsekwencji uszkodzenia przez energię promieniowania jest oparzenie „prętów” i „stożków” oka.
Na ryc. 1,43 pokazuje zależność i od m, a także T od m, co określa obszary znośnego i nie do zniesienia bólu podczas powstawania oparzeń termicznych w różnym stopniu. Kryterium zaimplementowane na rysunku opiera się na fakcie, że przy promieniowaniu cieplnym ból nie do zniesienia pojawia się, gdy temperatura warstwy skóry o grubości około 0,14-0,15 mm (pod powierzchnią górnej warstwy nabłonka) osiąga lub przekracza temperaturę 45° C.
Po wyeliminowaniu promieniowania (ale nie więcej niż 20-30 s) ostry ból ustępuje, a następnie z reguły całkowicie zanika. Wzrost temperatury określonej warstwy o 4-10 stopni lub więcej powoduje szok bólowy i oczywiste oparzenia skóry.
Obszar bólu znośnego pokazany na wykresie określa fakt, że w momencie narażenia na promieniowanie dochodzi do biologicznego odruchu ochronnego, powodującego wzrost przepływu krwi z obwodowych części ciała, co zapobiega miejscowemu wzrostowi w temperaturze do poziomu progowego. Pod wpływem wysokiej dawki ciśnienia termicznego ten fizjologiczny mechanizm nie jest już w stanie zapewnić niezbędnego odprowadzania ciepła, a organizm podlega patologicznym, a czasem zaporowym obciążeniom termicznym. Z natury linii na ryc. 1,42 pokazuje, że istnieje pewna ilościowa
dawka promieniowania q i temperatura T, która powoduje uszkodzenie termiczne i wystąpienie bólu nie do zniesienia, gdy ta dawka jest podana z niezbędnym czasem ekspozycji.
Czas ekspozycji, s Rys 1.43. Granice obrażeń termicznych i lekkich
Wypadki z samolotami (LA) zdarzają się głównie na skutek awarii jednostek, przede wszystkim - awarii silnika, ataków terrorystycznych, pożaru, a towarzyszą im wybuchy. Wybuch może nastąpić w powietrzu lub przy uderzeniu w ziemię. Gdy samolot spada na tereny mieszkalne, mogą ulec uszkodzeniu ludzie, konstrukcje itp. Przykłady katastrof lotniczych, ich analiza są podane w pracach.
Jednym z głównych zagrożeń w lotnictwie jest możliwość pożaru podczas awaryjnego lądowania. Paliwo wydostające się z uszkodzonych zbiorników może zapalić się od iskier ciernych z gorącego
powierzchni lub otwartego ognia. W takim przypadku powstałe centrum spalania szybko rozprzestrzenia się na wszystkie strefy, w których stosunki pary/paliwa do powietrza mieszczą się w zakresie palności. Jedną z metod zmniejszenia ryzyka pożaru jest stosowanie zżelowanych paliw, które rozprzestrzeniają się wolniej i są mniej lotne niż konwencjonalne paliwa płynne. Uszkodzenie zbiornika z zagęszczonym paliwem powoduje gwałtowne zmniejszenie zarówno szybkości rozprzestrzeniania się paliwa, jak i powstawania aerozoli palnych. Pozwala to wydłużyć czas, w którym pasażerowie mogą być ewakuowani.
Sytuacje nadzwyczajne i nadzwyczajne powodują ogromne szkody materialne i zaostrzają problemy środowiskowe. W przypadku wypadków, którym towarzyszą wybuchy i pożary, istnieje silny wpływ mechaniczny, termiczny i chemiczny na środowisko. Jednocześnie gwałtownie wzrastają emisje zanieczyszczeń; powierzchnia ziemi jest zaśmiecona fragmentami LL, pozostałościami paliwa, produktami spalania; znaczne szkody wyrządzono w naturalnym krajobrazie, florze, faunie; umierają pastwiska i żyzne gleby.
Uderzenie mechaniczne charakteryzuje się naruszeniem górnej (żyznej) warstwy gleby ze względu na jej powierzchnię i głębokie zniszczenie, wpływ energii wybuchu (fala uderzeniowa); naruszenie szaty trawiastej, uszkodzenie lub zamieranie krzewów, drzew i innej roślinności. Zmienia się struktura górnej warstwy żyznej, wymiana gazowo-wodna oraz struktura kapilarna.
Środki mające na celu poprawę bezpieczeństwa w sytuacjach awaryjnych zwykle dzielą się na dwie kategorie. Pierwsza obejmuje działania prowadzone po pojawieniu się
sytuacja awaryjna. Działania El1 nazywane są zwykle operacyjnymi i zasadniczo sprowadzają się do ochrony ludności i eliminowania skutków sytuacji kryzysowych. Druga grupa działań to działania realizowane z wyprzedzeniem. Obejmują one zwiększenie niezawodności urządzeń procesowych, zmniejszenie zapasów substancji niebezpiecznych w obiektach, usunięcie niebezpiecznego obiektu i podjęcie wczesnych działań w celu ochrony ludzi.
Duże znaczenie ma aktywny system bezpieczeństwa lotu (ASOSPS), który jest elementem pokładowego „inteligentnego” systemu wsparcia pilota, zwanego w praktyce lotniczej „asystentem pilota”, przeznaczonym do pracy zarówno w normalnych, jak i nienormalnych sytuacjach lotniczych . ASOBP wydaje sygnały ostrzegawcze o zagrożeniu bezpieczeństwa lotu, a także niezwłocznie informuje w formie „wskazówek” dotyczących sterowania samolotem i jego zespołem pokładowym, aby uniemożliwić samolotowi wejście w krytyczne tryby lotu. Aby zapobiec kolizjom z powierzchnią ziemi i między samolotami, ASOBP generuje przestrzenne trajektorie „rozrodu”.
Jednym z efektywnych obszarów pracy nad zapobieganiem wypadkom lotniczym jest pełne, dogłębne i obiektywne badanie zdarzeń, które już zaszły i wypracowanie na tej podstawie zaleceń zapobiegających ich powtórzeniu się.
Skuteczność takiej pracy zależy nie tylko od wystarczającego poziomu zasobów, ale także od wyczerpujących uprawnień organu prowadzącego niezależne dochodzenie, pozwalających mu wpływać na dowolny obszar systemu transportu lotniczego (produkcja, projektowanie, testowanie, certyfikacja , eksploatacja, naprawa, ramy regulacyjne itp.) .
Standardowy 5.4. Załącznik 13 do Konwencji o międzynarodowym lotnictwie cywilnym stanowi: „Organ badania wypadków uzyskuje niezależność w prowadzeniu badania i nieograniczone uprawnienia do jego prowadzenia”. Wymóg ten jest również uwzględniony w rosyjskich zasadach śledczych zatwierdzonych przez rząd Federacji Rosyjskiej. Utworzony na mocy umowy Międzypaństwowy Komitet Lotniczy (IAC) otrzymał od szefów państw i rządów WNP prawo do niezależnego badania wypadków lotniczych. Od 1992 roku specjaliści MAK zbadali ponad 270 wypadków lotniczych, w tym ponad 50 międzynarodowych, w tym badania zdarzeń z udziałem samolotów zachodnich.
Obecnie na świecie istnieje siedem takich wyspecjalizowanych ośrodków badania wypadków (USA, Francja, Wielka Brytania, Kanada, Niemcy, Australia i IAC).
Nie bez znaczenia jest dostarczanie państwom informacji o awariach i niesprawnościach sprzętu lotniczego oraz błędnych działaniach załóg. Korzystając z tych danych, władze lotnicze każdego państwa mogą podjąć środki zapobiegawcze.