Processus isobare dans le diagramme ts. Utilisation des diagrammes d'état TS, Pv et hs de la vapeur d'eau dans les calculs techniques. a) Processus isochore
Lire aussi
La figure 3.3 montre le diagramme de phase en coordonnées P – V et la figure 3.4 – en coordonnées T – S.

Figure 3.3. Diagramme PV de phase Fig. 3.4. Phase Diagramme TS
Désignations:
t + l – région de coexistence à l’équilibre du solide et du liquide
t + p – région de coexistence à l'équilibre du solide et de la vapeur
l + n – région de coexistence à l’équilibre du liquide et de la vapeur
Si sur le diagramme P – T les zones des états biphasés étaient représentées sous forme de courbes, alors les diagrammes P – V et T – S sont des zones.
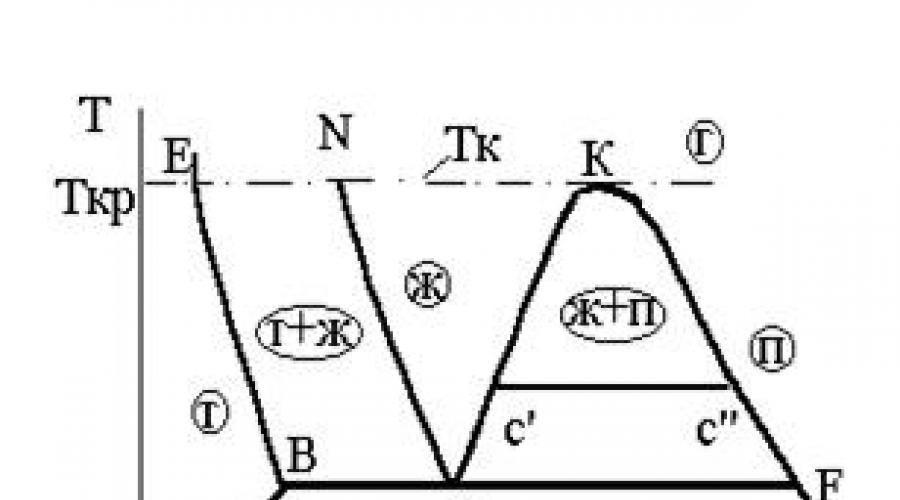
La ligne AKF est appelée courbe limite. Elle est à son tour divisée en une courbe limite inférieure (section AK) et une courbe limite supérieure (section KF).
Sur les figures 3.3 et 3.4, la ligne BF, où se rencontrent les régions de trois états biphasés, est le point triple étendu T des figures 3.1 et 3.2.
Lorsqu'une substance fond, ce qui, comme la vaporisation, se produit à température constante, un mélange biphasique d'équilibre de phases solides et liquides se forme. Les valeurs du volume spécifique de la phase liquide dans la composition d'un mélange diphasique sont tirées de la figure 3.3 à partir de la courbe AN, et les valeurs du volume spécifique de la phase solide - de la courbe BE .
A l'intérieur de la zone délimitée par le contour AKF, la substance est un mélange de deux phases : liquide bouillant (L) et vapeur sèche saturée (P).
En raison de l'additivité du volume, le volume spécifique d'un tel mélange biphasique est déterminé par la formule
![]()
entropie spécifique :
![]()
Points spéciaux diagrammes de phases
Point triple
Le point triple est le point où convergent les courbes d’équilibre des trois phases. Sur les figures 3.1 et 3.2, il s'agit du point T.
Certaines substances pures, par exemple le soufre, le carbone, etc., sous forme solide état d'agrégation comporte plusieurs phases (modifications).
Il n’y a aucune modification aux états liquide et gazeux.
Conformément à l'équation (1.3), dans un système de déformation thermique à un composant, pas plus de trois phases ne peuvent être simultanément en équilibre.
Si une substance présente plusieurs modifications à l’état solide, alors total Les phases d'une substance dépassent au total trois et une telle substance doit avoir plusieurs points triples. À titre d'exemple, la figure 3.5 montre le diagramme de phase P – T d'une substance qui présente deux modifications à l'état solide d'agrégation.

Figure 3.5. Diagramme phase P-T
substances à deux cristaux
quelles phases
Désignations:
I – phase liquide ;
II – phase gazeuse ;
III 1 et III 2 – modifications à l’état solide d’agrégation
(phases cristallines)
Au point triple T 1, sont en équilibre : la phase gazeuse, liquide et cristalline III 2. Ce point est basique point triple.
Au point triple T2, sont en équilibre : les phases liquide et deux phases cristallines.
Au point triple T3, les phases gazeuse et cristalline sont en équilibre.
Il existe cinq modifications (phases) cristallines connues de l'eau : III 1, III 2, III 3, III 5, III 6.
La glace ordinaire est la phase cristalline III 1, et d'autres modifications se forment à des pressions très élevées de plusieurs milliers de MPa.
La glace ordinaire existe jusqu'à une pression de 204,7 MPa et une température de 22 0 C.
Les modifications (phases) restantes sont la glace plus dense que l'eau. L'une de ces glaces, « glace chaude », a été observée à une pression de 2000 MPa jusqu'à une température de + 80 0 C.
Paramètres thermodynamiques eau de base à triple point ce qui suit:
T tr = 273,16 K = 0,01 0 C ;
P tr = 610,8 Pa ;
Vtr = 0,001 m 3 /kg.
L'anomalie de la courbe de fusion () n'existe que pour la glace ordinaire.
Point critique
Comme il ressort du diagramme de phase P – V (Fig. 3.3), à mesure que la pression augmente, la différence entre les volumes spécifiques de liquide bouillant (V") et de vapeur sèche saturée (V"") diminue progressivement et au point K devient égal à zéro. Cet état est appelé critique , et le point K est le point critique de la substance.
Pk, Tk, Vk, Sk – paramètres thermodynamiques critiques de la substance.
Par exemple, pour l'eau :
Pk = 22,129 MPa ;
Tc = 374,14 0 C ;
V k = 0,00326 m 3 /kg
Au point critique, les propriétés des phases liquide et gazeuse sont les mêmes.
Comme le montre le diagramme de phase T – S (Figure 3.4), au point critique, la chaleur de vaporisation, représentée comme la zone située sous la ligne de transition de phase horizontale (C" - C""), du liquide bouillant à la vapeur saturée sèche, est zéro.
Le point K de l'isotherme Tk dans le diagramme de phases P – V (Fig. 3.3) est un point d'inflexion.
L'isotherme Tk passant par le point K est ultime isotherme de la région biphasée, c'est-à-dire sépare la région de la phase liquide de la région gazeuse.
Aux températures supérieures à Tk, les isothermes n'ont plus de sections droites indiquant des transitions de phase, ni le point d'inflexion caractéristique de l'isotherme Tk, mais prennent progressivement la forme de courbes lisses, de forme proche des isothermes d'un gaz parfait.
Les notions de « liquide » et de « gaz » (vapeur) dans dans une certaine mesure conditionnel, parce que les interactions des molécules dans le liquide et le gaz ont modèles généraux, ne différant que quantitativement. Cette thèse peut être illustrée par la figure 3.6, où la transition du point E de la phase gazeuse au point L de la phase liquide se fait en contournant le point critique K le long de la trajectoire EFL.

Figure 3.6. Options de transition en deux phases
de la phase gazeuse à la phase liquide
En passant le long de la ligne AD au point C, la substance se sépare en deux phases puis la substance passe progressivement de la phase gazeuse (vapeur) à la phase liquide.
Au point C, les propriétés de la substance changent brusquement (dans le diagramme de phase P – V, le point C de la transition de phase se transforme en une ligne de transition de phase (C" - C")).
Lors du déplacement le long de la ligne EFL, la transformation du gaz en liquide se produit en continu, puisque la ligne EFL ne coupe nulle part la courbe de vaporisation du TC, où la substance existe simultanément sous la forme de deux phases : liquide et gazeuse. Par conséquent, lors du passage sur la ligne EFL, la substance ne se décomposera pas en deux phases et restera monophasée.
Température critique Tc est la température limite pour la coexistence à l’équilibre de deux phases.
En ce qui concerne les processus thermodynamiques dans systèmes complexes Cette définition laconique classique de T k peut être développée comme suit :
Température critique Tc - il s'agit de la limite inférieure de température de la région des processus thermodynamiques dans laquelle l'apparition d'un état biphasé d'une substance « gaz - liquide » est impossible sous tout changement de pression et de température. Cette définition est illustrée dans les figures 3.7 et 3.8. De ces chiffres, il résulte que cette zone est limitée température critique, couvre uniquement l'état gazeux de la substance (phase gazeuse). L’état gazeux de la substance, appelé vapeur, n’est pas inclus dans cette région.


Riz. 3.7. Vers la définition du critique Fig. 3.8. Vers la définition du critique
température
Il résulte de ces figures que cette zone grisée, limitée par la température critique, ne couvre que l'état gazeux de la substance (phase gazeuse). L’état gazeux de la substance, appelé vapeur, n’est pas inclus dans cette région.
En utilisant la notion de point critique, on peut concept général« état gazeux de la matière » met en avant la notion de « vapeur ».
Vapeur – il s’agit de la phase gazeuse d’une substance dans la plage de température inférieure à la température critique.
Dans les procédés thermodynamiques, lorsque la ligne de procédé croise soit la courbe de vaporisation TC, soit la courbe de sublimation 3, la phase gazeuse est toujours initialement vapeur.
Pression critique P k - c'est la pression au-dessus de laquelle la séparation d'une substance en deux phases coexistant simultanément et à l'équilibre : liquide et gaz est impossible à n'importe quelle température.
Cette définition classique de P k, en relation avec les processus thermodynamiques dans des systèmes complexes, peut être formulée plus en détail :
Pression critique P k - il s'agit de la limite de pression inférieure de la région des processus thermodynamiques dans laquelle l'apparition d'un état biphasique d'une substance « gaz - liquide » est impossible sous tout changement de pression et de température. Cette définition de la pression critique est illustrée sur la figure 3.9. et 3.10. Il résulte de ces figures que cette région, limitée par la pression critique, couvre non seulement la partie de la phase gazeuse située au dessus de l'isobare Pk, mais également la partie de la phase liquide située en dessous de l'isotherme Tk.
Pour la région supercritique, l'isotherme critique est classiquement considérée comme la limite liquide-gaz probable (conditionnelle).


Fig. 3.9. Vers la définition du critique - Fig. 3.10. Vers la définition de la critique
qui est la pression de la pression
Si la pression de transition est bien supérieure à la pression au point critique, alors la substance passera directement de l'état solide (cristallin) à l'état gazeux, en contournant état liquide.
Cela n'est pas évident d'après les diagrammes de phases P-T de la substance anormale (Figures 3.6, 3.7, 3.9), car ils ne montrent pas la partie du diagramme où une substance qui, à haute pression, présente plusieurs modifications cristallines (et, par conséquent, plusieurs points triples), acquiert à nouveau des propriétés normales.
Sur le diagramme phase P – T de la matière normale, Fig. 3.11 ce passage de la phase solide directement à la phase gazeuse est représenté sous la forme du procédé A "D".

Riz. 3.11. Transition vers la normale
substances de la phase solide immédiatement dans
gazeux à P>Ptr
Le passage d'une substance de la phase solide à la phase vapeur, en contournant la phase liquide, n'est attribué qu'à P<Р тр. Примером такого перехода, называемого сублимацией, является процесс АD на рис 3.11.
La température critique a une interprétation cinétique moléculaire très simple.
La combinaison de molécules en mouvement libre dans une goutte de liquide lors de la liquéfaction du gaz se produit uniquement sous l'influence de forces d'attraction mutuelle. Quand T>T k énergie cinétique le mouvement relatif de deux molécules est supérieur à l'énergie d'attraction de ces molécules, donc la formation de gouttelettes liquides (c'est-à-dire la coexistence de deux phases) est impossible.
Seules les courbes de vaporisation présentent des points critiques, puisqu'elles correspondent à la coexistence à l'équilibre de deux isotrope phases : liquide et gazeuse. Les lignes de fusion et de sublimation n'ont pas de points critiques, car ils correspondent à de tels états biphasiques de la matière, lorsqu'une des phases (solide) est anisotrope.
Région supercritique
Dans un diagramme de phase P-T, il s'agit de la région située à droite et au-dessus du point critique, à peu près là où l'on pourrait mentalement continuer la courbe de saturation.
En flux direct moderne chaudières à vapeur la vaporisation se produit dans la région supercritique.


Figure 3.12. Transition de phase sur la figure 3.13. Transition de phase en sous-critique
sous-critique et supercritique et supercritique zones P-V diagrammes
régions RT diagrammes
Les processus thermodynamiques dans la région supercritique se produisent avec un certain nombre de caractéristiques distinctives.
Considérons le processus isobare AS dans la région sous-critique, c'est-à-dire à . Le point A correspond à la phase liquide de la substance qui, lorsque la température Tn est atteinte, commence à se transformer en vapeur. Cette transition de phase correspond au point B de la figure 3.12 et au segment B"B"" de la figure 3.13. Lors du passage par la courbe de saturation TK, les propriétés de la substance changent brusquement. Le point S correspond à la phase gazeuse de la substance.
Considérons le processus isobare A"S" à pression . Au point A", la substance est en phase liquide, et au point S" - en phase gazeuse, c'est-à-dire dans différents états de phase. Mais lorsqu’on passe du point A" au point S", il n’y a pas de changement brusque des propriétés : les propriétés de la substance changent continuellement et progressivement. Le taux de ce changement des propriétés de la substance sur la ligne A"S" est différent : il est faible près des points A" et S" et augmente fortement à l'entrée dans la région supercritique. Sur n'importe quelle isobare de la région supercritique, vous pouvez indiquer des points vitesse maximum changements : coefficient de température de dilatation volumétrique d'une substance, enthalpie, énergie interne, viscosité, conductivité thermique, etc.
Ainsi, dans la région supercritique, des phénomènes similaires à des transitions de phase se développent, mais l'état biphasique de la substance « liquide - gaz » n'est pas observé. De plus, les limites de la région supercritique sont floues.
À P<Р к, т.е. в докритической области, на фазовое превращение «жидкость - пар» требуется затратить скрытую теплоту парообразования, которая является как бы «тепловым барьером» между жидкой и паровой фазами.
Quelque chose de similaire est observé dans la région supercritique. La figure 3.14 montre une image typique des changements dans la capacité thermique isobare spécifique à P>P k.

Figure 3.14. Isobare spécifique
capacité thermique à supercritique
pression.
Puisque Q р = С р dТ, l'aire sous la courbe Ср(Т) est la chaleur nécessaire pour convertir le liquide (point A') en gaz (point S') à pression supercritique. La ligne pointillée А'М S' montre une dépendance typique de Ср à la température dans sous-critique zones.
Ainsi, les maxima sur la courbe C p (T) dans la région supercritique, c'est-à-dire la consommation de chaleur supplémentaire pour chauffer la substance, remplissent également des fonctions similaires en tant que « barrière thermique » entre le liquide et le gaz dans cette région.
Comme les études l'ont montré, les positions des maximums  ne coïncident pas, ce qui indique l'absence d'une seule ligne entre le liquide et la vapeur dans la région supercritique. Il n'y a qu'une zone large et floue, où la transformation du liquide en vapeur se produit le plus intensément.
ne coïncident pas, ce qui indique l'absence d'une seule ligne entre le liquide et la vapeur dans la région supercritique. Il n'y a qu'une zone large et floue, où la transformation du liquide en vapeur se produit le plus intensément.
Ces transformations se produisent le plus intensément à des pressions qui ne dépassent pas la pression critique (Pc). Au fur et à mesure que la pression augmente, les phénomènes de transformation du liquide en vapeur s'estompent et à haute pression apparaissent très faiblement.
Ainsi, à P>P k il y a, mais ne peuvent coexister simultanément et à l'équilibre, une phase liquide, une phase gazeuse et une phase intermédiaire. Cette phase intermédiaire est parfois appelée métaphase , il combine les propriétés du liquide et du gaz.
En raison du changement brutal des paramètres thermodynamiques, des caractéristiques thermophysiques et des fonctions caractéristiques dans la région supercritique, leurs erreurs détermination expérimentale dans cette région est plus de dix fois supérieure à celle des pressions sous-critiques.
Isoprocédés des gaz parfaits– les processus dans lesquels l'un des paramètres reste inchangé.
1. Processus isochore . La loi de Charles. V = const.
Processus isochore appelé un processus qui se produit lorsque volume constant V. Le comportement du gaz dans ce processus isochore obéit La loi de Charles :
A volume constant et valeurs constantes de la masse de gaz et de sa masse molaire, le rapport de la pression du gaz à sa température absolue reste constant : P/T= const.
Graphique d'un processus isochore sur PV-le diagramme s'appelle isochore . Il est utile de connaître le graphique d'un processus isochore sur RT- Et Vermont-des diagrammes (Fig. 1.6). Équation isochore :
Où P 0 est la pression à 0 °C, α est le coefficient de température de la pression du gaz égal à 1/273 deg -1. Un graphique d'une telle dépendance à Рt-le diagramme a la forme montrée sur la figure 1.7.

Riz. 1.7
2. Processus isobare. Loi de Gay-Lussac. R.= const.
Un processus isobare est un processus qui se produit à pression constante P . Le comportement d'un gaz au cours d'un processus isobare obéit Loi de Gay-Lussac:
A pression constante et valeurs constantes de la masse du gaz et de sa masse molaire, le rapport du volume du gaz à sa température absolue reste constant : VERMONT= const.
Graphique d'un processus isobare sur Vermont-le diagramme s'appelle isobare . Il est utile de connaître les graphiques du processus isobare sur PV- Et RT-des diagrammes (Fig. 1.8).

Riz. 1.8
Équation isobare :
Où α =1/273 deg -1 - coefficient de température de dilatation volumétrique. Un graphique d'une telle dépendance à Vermont Le diagramme a la forme illustrée à la figure 1.9.

Riz. 1.9
3. Processus isotherme. Loi Boyle-Mariotte. T= const.
Isotherme le processus est un processus qui se produit lorsque Température constante T.
Le comportement d'un gaz parfait lors d'un processus isotherme obéit Loi Boyle-Mariotte :
A température constante et valeurs constantes de la masse du gaz et de sa masse molaire, le produit du volume du gaz et de sa pression reste constant : PV= const.
Graphique d'un processus isotherme sur PV-le diagramme s'appelle isotherme . Il est utile de connaître les graphiques d'un processus isotherme sur Vermont- Et RT-des diagrammes (Fig. 1.10).

Riz. 1.10
Équation isotherme :
| (1.4.5) |
4. Processus adiabatique( isentropique ) :
Le processus adiabatique est un processus thermodynamique qui se produit sans échange de chaleur avec environnement.
5. Processus polytropique. Processus dans lequel la capacité thermique d'un gaz reste constante. Le processus polytropique est un cas général de tous les processus énumérés ci-dessus.
6. La loi d'Avogadro. Aux mêmes pressions et aux mêmes températures, des volumes égaux de différents gaz parfaits contiennent même nombre molécules. Dans un centre commercial diverses substances contient N A=6,02·10 23 molécules (numéro d'Avogadro).
7. La loi de Dalton. La pression d'un mélange de gaz parfaits est égale à la somme des pressions partielles P des gaz qu'il contient :
| (1.4.6) |
La pression partielle Pn est la pression qu'exercerait un gaz donné s'il occupait à lui seul tout le volume.
À ![]() , pression du mélange gazeux.
, pression du mélange gazeux.
Phase pv – diagramme système composé de liquide et de vapeur, est un graphique des volumes spécifiques d’eau et de vapeur en fonction de la pression.
Laisser l'eau à température 0 0 C et une certaine pression ρ occupe un volume spécifique v 0 (segment N.-É.) . Toute la courbe AE exprime la dépendance du volume spécifique d'eau sur la pression à la température 0 0 C. Parce que l'eau est une substance presque incompressible qui est une courbe AE presque parallèle à l'axe des ordonnées. Si de la chaleur est transmise à l’eau à pression constante, sa température augmentera et son volume spécifique augmentera. A une certaine température ts l'eau bout et son volume spécifique v'à ce point UN' atteindra sa valeur maximale à une pression donnée. À mesure que la pression augmente, la température du liquide bouillant augmente ts et le volume v' augmente également. Graphique de dépendance v' en fonction de la pression est représenté par une courbe AK qui est appelée la courbe limite fluide. La caractéristique de la courbe est le degré de sécheresse x=0. En cas d'apport de chaleur supplémentaire à pression constante, le processus de vaporisation commencera. Dans le même temps, la quantité d'eau diminue, la quantité de vapeur augmente. Au moment de l'achèvement de la vaporisation au point DANS' la vapeur sera sèche et saturée. Le volume spécifique de vapeur saturée sèche est désigné v''.
Si le processus de vaporisation se produit à pression constante, alors sa température ne change pas et le processus UN B' est à la fois isobare et isotherme. Aux points UN' Et B' la substance est dans un état monophasé. Aux points intermédiaires, la substance est constituée d'un mélange d'eau et de vapeur. Ce mélange de corps s'appelle système biphasé.
Tracé de volume spécifique v'' en fonction de la pression est représenté par une courbe KV, qui est appelée courbe limite de vapeur.
Si de la chaleur est fournie à de la vapeur saturée sèche à pression constante, alors sa température et son volume augmenteront et la vapeur saturée sèche passera de saturée sèche à surchauffée (point D). Les deux courbes AK Et HF divisez le diagramme en trois parties. À gauche de la courbe limite du fluide AK Avant l’isotherme zéro, il y a une région liquide. Entre les courbes AK Et HF il existe un système biphasé constitué d'un mélange d'eau et de vapeur sèche. Directement de HF et à partir du point À il existe une zone de vapeur surchauffée ou d'état gazeux du corps. Les deux courbes AK Et HF converger en un point À, appelé le point critique.
Le point critique est le point final de la transition de phase liquide-vapeur commençant au point triple. Au-dessus du point critique, l’existence d’une substance dans un état biphasique est impossible. Aucune pression ne peut transformer un gaz à l’état liquide à des températures supérieures à la valeur critique.
Paramètres des points critiques pour l’eau :
tk =374,12 0 C ; v k = 0,003147 m 3 /kg;
ρ k = 22,115 MPa ; je k =2095,2 kJ/kg
sk =4,424 kJ/(kgK).
Processus p = const p-V , est Et T-S des diagrammes.

Sur est – diagramme L'isobare dans la région de la vapeur saturée est représentée par une ligne droite coupant les courbes limites de la vapeur liquide. Lorsque de la chaleur est fournie à la vapeur humide, son degré de sécheresse augmente et elle (à température constante) se transforme en vapeur sèche et, avec un apport de chaleur supplémentaire, en vapeur surchauffée. L'isobare dans la région de la vapeur surchauffée est une courbe dirigée de manière convexe vers le bas.

Sur pv – diagramme un processus isobare est représenté par un segment de ligne droite horizontale qui, dans la zone de la vapeur humide, représente également en même temps un processus isotherme.

Sur Ts – diagramme dans la région de la vapeur humide, l'isobare est représentée par une ligne droite horizontale, et dans la région de la vapeur surchauffée - par une courbe convexe vers le bas. Les valeurs de toutes les grandeurs nécessaires au calcul sont tirées des tableaux des vapeurs saturées et surchauffées.
Modification de l'énergie interne spécifique de la vapeur :
Quantité spécifique de chaleur fournie :
Dans le cas où qétant donné et il faut trouver les paramètres du deuxième point se situant dans la région des états diphasiques, la formule de l'enthalpie de la vapeur humide est appliquée :
Processus T=const vapeur d'eau. Image de processus dans p-V , est Et T-S des diagrammes.
Processus isotherme.

Sur est – diagramme dans la région de la vapeur humide, l'isotherme coïncide avec l'isobare et est une ligne droite inclinée. Dans la région de la vapeur surchauffée, l’isotherme est représentée par une courbe à convexité ascendante.
1) En thermodynamique, ils sont largement utilisés pour étudier les processus d’équilibre. PV– un diagramme dans lequel l'axe des abscisses est le volume spécifique et l'axe des ordonnées est la pression. Puisque l’état d’un système thermodynamique est déterminé par deux paramètres, alors PV– dans le diagramme, il est représenté par un point. Sur la figure, le point 1 correspond à l'état initial du système, le point 2 à l'état final et la ligne 1-2 au processus de détente du fluide de travail de v 1 à v 2. Pour un changement de volume infinitésimal dv l'aire de la bande verticale ombrée est égale à pdv = δl, par conséquent, le travail du processus 1-2 est représenté par la zone limitée par la courbe du processus, l'axe des x et les ordonnées extrêmes. Ainsi, le travail de changement de volume est équivalent à l'aire sous la courbe de processus dans le diagramme PV.

2) L'état d'équilibre dans le diagramme TS est représenté par des points dont les coordonnées correspondent aux valeurs de température et d'entropie. Dans ce diagramme, la température est tracée le long de l’axe des ordonnées et la température est tracée le long de l’axe des abscisses. entropie.
Le processus thermodynamique réversible de changement de l'état du fluide de travail de l'état initial 1 à l'état final 2 est représenté sur le diagramme TS par une courbe continue passant entre ces points. L'aire abdc est égale à TdS=dq, c'est-à-dire exprime la quantité élémentaire de chaleur reçue ou dégagée par un système dans un processus réversible. Aire sous la courbe en TS− Le diagramme représente la chaleur fournie ou évacuée du système. C'est pourquoi TS− Le diagramme est appelé diagramme thermique.

Procédés gazeux dans le diagramme TS−.
1. Processus isotherme.
Dans un processus isotherme T=const. C'est pourquoi T.S.−dans le diagramme, il est représenté par une ligne droite parallèle à l'axe des abscisses.
2. Processus adiabatique
Dans un processus adiabatique q=0 Et dq=0, et par conséquent dS=0.
Donc dans un processus adiabatique S=const et en T.S.−dans le diagramme, le processus adiabatique est représenté par une ligne droite parallèle à l'axe T. Puisque dans un processus adiabatique S=const, alors les processus adiabatiques réversibles sont également appelés isentropiques. Lors de la compression adiabatique, la température du fluide de travail augmente et lors de la détente, elle diminue. Par conséquent, le processus 1-2 est un processus de compression et le processus 2-1 est un processus d'expansion.
Pour un processus isochore V=const, dV=0.À capacité thermique constante - vue de T.S.-diagramme. La sous-tangente à la courbe de processus détermine en tout point la valeur de la véritable capacité thermique CV. La sous-tangente ne sera positive que si la courbe est convexe vers le bas.
4. Processus isobare
Dans un processus isobare, la pression est constante p=const.
À p=const comme avec V=const Une isobare est une courbe logarithmique, montant de gauche à droite et convexe vers le bas.
La sous-tangente à la courbe 1-2 donne en tout point les valeurs de la véritable capacité thermique Cp.
Processus thermodynamique (processus thermique) – un changement dans l’état macroscopique d’un système thermodynamique. Si la différence entre les états initial et final du système est infinitésimale, alors un tel processus est appelé élémentaire (infinitésimal).
Le système dans lequel se produit le processus thermique est appelé fluide de travail.
Les processus thermiques peuvent être divisés en équilibre et hors équilibre. Un processus d’équilibre est un processus dans lequel tous les états par lesquels passe le système sont des états d’équilibre. Un tel processus est approximativement réalisé dans les cas où les changements se produisent plutôt lentement, c'est-à-dire que le processus est quasi statique.
Les processus thermiques peuvent être divisés en réversibles et irréversibles. Réversible est un processus qui peut s'effectuer dans le sens opposé à travers tous les mêmes états intermédiaires.
Types de procédés thermiques :
Processus adiabatique – sans échange thermique avec l’environnement. environnement;
Processus isochore - se produisant à volume constant ;
Processus isobare - se produisant à pression constante ;
Processus isotherme - se produisant à température constante ;
Processus isoentropique - se produisant à entropie constante ;
Processus isenthalpique - se produisant à enthalpie constante ;
Processus polytropique - se produisant à capacité thermique constante.
Équation de Mendeleïev-Clayperon (équation d'état des gaz parfaits) :
PV = nRT, où n est le nombre de moles de gaz, P est la pression du gaz, V est le volume du gaz, T est la température du gaz, R est la constante universelle des gaz.
Isoprocessus d'un gaz parfait. Leur image dans P. - V des diagrammes.
1) Processus isobare p = const, V/T = const



2) Processus isochore V = const, p/T = const



3) Processus isotherme T = const, pV = const



Processus thermodynamiques. Équation de Mendeleïev-Clapeyron. Isoprocessus d'un gaz parfait. Leur image sur R-Vdes diagrammes.
Processus thermodynamiques. L’ensemble des états changeants du fluide de travail est appelé processus thermodynamique.
Un gaz parfait est un gaz imaginaire étudié en thermodynamique, dans lequel il n'y a pas de forces d'attraction et de répulsion intermoléculaires, et les molécules elles-mêmes sont points matériels, n'ayant pas de volume. De nombreux gaz réels sont très proches dans leurs propriétés physiques d’un gaz parfait.
Les principaux processus en thermodynamique sont :
isochore, circulant à volume constant ;
isobare, circulant à pression constante ;
isotherme, se produisant à température constante ;
adiabatique, dans lequel il n'y a pas d'échange thermique avec l'environnement ;
Processus isochore

Dans un processus isochore, la condition est satisfaite v= const.
De l'équation d'état d'un gaz parfait ( PV=RT) suit :
p/T=RV= const,
c'est-à-dire que la pression du gaz est directement proportionnelle à sa température absolue :
p 2 /p 1 =T 2 /T 1 .
Le travail d’expansion dans un processus isochore est nul ( je= 0), puisque le volume du fluide de travail ne change pas (Δ v= const).
La quantité de chaleur fournie au fluide de travail dans le processus 1-2 à cv
q=cv(T. 2 -T 1 ).
Parce que je= 0, alors basé sur la première loi de la thermodynamique Δ toi=q, ce qui signifie que le changement d'énergie interne peut être déterminé par la formule :
Δ toi=cv(T. 2 -T 1 ).
Le changement d'entropie dans un processus isochore est déterminé par la formule :
s 2 -s 1 = Δ s = cv ln( p 2 /p 1 ) = cv ln( T 2 /T 1 ).
Processus isobare

Un processus qui se produit à pression constante est appelé isobare. p= const. De l’équation d’état d’un gaz parfait il résulte :
v/ T=R./ p=const
v 2 /v 1 =T 2 /T 1 ,
c'est-à-dire que dans un processus isobare, le volume d'un gaz est proportionnel à sa température absolue.
Le travail sera égal à :
je=p(v 2 –v 1 ).
Parce que PV 1 =RT 1 Et PV 2 =RT 2 , Que
je=R.(T 2 – T. 1 ).
Quantité de chaleur à cp= const est déterminé par la formule :
q=cp(T 2 – T. 1 ).
Le changement d'entropie sera égal à :
s 2 -s 1 = Δ s = cp ln( T 2 /T 1 ).
Processus isotherme

Dans un procédé isotherme, la température du fluide de travail reste constante T= const, donc :
PV = RT= const
p 2 / p 1 =v 1 / v 2 ,
c'est-à-dire que la pression et le volume sont inversement proportionnels l'un à l'autre, de sorte que pendant la compression isotherme, la pression du gaz augmente et pendant la détente, elle diminue.
Le travail du processus sera égal à :
je=RT ln( v 2 –v 1 ) =RT ln( p 1 -p 2 ).
Puisque la température reste constante, l'énergie interne d'un gaz parfait dans un processus isotherme reste constante (Δ toi= 0) et toute la chaleur fournie au fluide de travail est entièrement convertie en travail de détente :
q=l.
Lors de la compression isotherme, la chaleur est évacuée du fluide de travail en quantité égale au travail dépensé en compression.
Le changement d'entropie est :
s 2 -s 1 = Δ s=R. ln( p 1 /p 2 ) =R. ln( v 2 /v 1 ).
Processus adiabatique

L'adiabatique est le processus de changement d'état d'un gaz qui se produit sans échange thermique avec l'environnement. Depuis d q= 0, alors l'équation de la première loi de la thermodynamique pour un processus adiabatique aura la forme :
d toi+p d v= 0
Δ toi+je= 0,
ainsi
Δ toi= -l.
Dans un processus adiabatique, le travail d'expansion est effectué uniquement en dépensant l'énergie interne du gaz, et lors de la compression, qui se produit sous l'action de forces externes, tout le travail effectué par celles-ci sert à augmenter l'énergie interne du gaz. .
Notons la capacité thermique dans un processus adiabatique par c l'enfer et la condition d q= 0 on l'exprime comme suit :
d q=c bon sang T= 0.
Cette condition indique que la capacité thermique dans un processus adiabatique est nulle ( c enfer = 0).
Il est connu que
Avecp/cv =k
et l'équation de la courbe du processus adiabatique (adiabatique) dans p, v-le schéma ressemble à :
PVk= const.
Dans cette expression k est appelé indice adiabatique(également appelé coefficient de Poisson).
Valeurs de l'indice adiabatique k pour certains gaz :
k air = 1,4
k vapeur surchauffée = 1,3
k gaz d'échappement des moteurs à combustion interne = 1,33
k vapeur humide saturée = 1,135
Des formules précédentes il résulte :
je= - Δ toi = cv(T 1 – T. 2 );
je 1 - je 2 = cp(T 1 – T. 2 ).
Travaux techniques du processus adiabatique ( je techn) est égal à la différence entre les enthalpies de début et de fin du processus ( je 1 - je 2 ).
Un processus adiabatique se produisant sans frottement interne dans le fluide de travail est appelé isentropique. DANS T, s-dans le diagramme, il est représenté par une ligne verticale.
Généralement, de véritables processus adiabatiques se produisent en présence de friction interne dans le fluide de travail, ce qui entraîne toujours un dégagement de chaleur qui est communiqué au fluide de travail lui-même. Dans ce cas d s> 0, et le processus est appelé véritable processus adiabatique.
Équation de Mendeleïev-Clapeyron
Les gaz sont souvent des réactifs et des produits réactions chimiques. Il n’est pas toujours possible de les faire réagir les uns avec les autres dans des conditions normales. Par conséquent, vous devez apprendre à déterminer le nombre de moles de gaz dans des conditions autres que la normale.
Pour cela, ils utilisent équation d'état des gaz parfaits(également appelée équation de Clapeyron-Mendeleev) :
PV = n RT
Où n– nombre de moles de gaz ;
P – pression du gaz (par exemple, dans au m;
V – volume de gaz (en litres) ;
T – température du gaz (en kelvins) ;
R – constante du gaz (0,0821 l au m/mol K).
Par exemple, dans un ballon de 2,6 litres, il y a de l'oxygène à une pression de 2,3 au m et une température de 26 o C. Question : combien de moles d'O 2 sont contenues dans le ballon ?
À partir de la loi des gaz, nous trouvons le nombre de moles requis n:
Il ne faut pas oublier de convertir la température des degrés Celsius en Kelvin : (273 o C + 26 o C) = 299 K. D'une manière générale, afin de ne pas se tromper dans de tels calculs, il faut surveiller attentivement la dimension des valeurs. substitué dans l'équation de Clapeyron-Mendeleev. Si la pression est donnée en mm Mercure, alors vous devez le convertir en atmosphères en fonction du rapport : 1 au m= 760 mmHg. Art. La pression donnée en pascals (Pa) peut également être convertie en atmosphères, sur la base du fait que 101325 Pa = 1 au m.
Billet 16
Dérivation de l'équation de base de la théorie de la cinétique moléculaire. Le nombre de degrés de liberté d'une molécule. Loi de répartition de l'énergie en degrés de liberté.
Dérivation de l'équation de base MKT.



Le nombre de degrés de liberté d'une molécule. Loi de répartition de l'énergie en degrés de liberté.


Billet 17.
La première loi de la thermodynamique. Le gaz fonctionne lorsque le volume change. Calculer le travail de dilatation isotherme du gaz.
Quantité de chaleur, reçu par le système, va changer son énergie interne et effectuer un travail contre les forces externes
La variation de l'énergie interne d'un système lors de sa transition d'un état à un autre est égale à la somme du travail des forces externes et de la quantité de chaleur transférée au système, c'est-à-dire qu'elle dépend uniquement de l'état initial et final. du système et ne dépend pas de la manière dont s’effectue cette transition. Dans un processus cyclique, l’énergie interne ne change pas.





Le travail pendant la dilatation isotherme d'un gaz est calculé comme l'aire de la figure sous le graphique de processus.


Billet 18.
Capacité thermique d'un gaz parfait.
Si, à la suite d'un échange de chaleur, une certaine quantité de chaleur est transférée au corps, alors l'énergie interne du corps et sa température changent. La quantité de chaleur Q nécessaire pour chauffer 1 kg d'une substance de 1 K est appelée capacité thermique spécifique de la substance c. c = Q / (mΔT).
où M est la masse molaire de la substance.
La capacité thermique ainsi déterminée n'est pas une caractéristique univoque d'une substance. Selon la première loi de la thermodynamique, la variation de l’énergie interne d’un corps dépend non seulement de la quantité de chaleur reçue, mais aussi du travail effectué par le corps. Selon les conditions dans lesquelles le processus de transfert de chaleur s'effectue, le corps peut effectuer divers travaux. Par conséquent, la même quantité de chaleur transférée à un corps pourrait provoquer différents changements dans son énergie interne et, par conséquent, dans sa température.
Cette ambiguïté dans la détermination de la capacité thermique n'est typique que pour les substances gazeuses. Lorsque des liquides et des solides sont chauffés, leur volume ne change pratiquement pas et le travail de dilatation s'avère nul. Par conséquent, toute la quantité de chaleur reçue par le corps sert à modifier son énergie interne. Contrairement aux liquides et solides, le gaz en cours de transfert de chaleur peut modifier considérablement son volume et effectuer un travail. Par conséquent, la capacité thermique d’une substance gazeuse dépend de la nature du processus thermodynamique. Habituellement, deux valeurs de la capacité thermique des gaz sont considérées : C V – capacité thermique molaire dans un processus isochore (V = const) et C p – capacité thermique molaire dans un processus isobare (p = const).
Dans le processus à volume constant, le gaz ne fait aucun travail : A = 0. De la première loi de la thermodynamique pour 1 mole de gaz il découle
où ΔV est la variation de volume de 1 mole d'un gaz parfait lorsque sa température change de ΔT. Cela implique:
où R est la constante universelle des gaz. Pour p = const
Ainsi, la relation exprimant le rapport entre les capacités thermiques molaires C p et C V a la forme (formule de Mayer) :
|
C p = C V + R. |
La capacité thermique molaire C p d'un gaz dans un procédé à pression constante est toujours supérieure à la capacité thermique molaire C V dans un procédé à volume constant
Le rapport des capacités thermiques dans les processus à pression constante et à volume constant joue un rôle important en thermodynamique. Il est désigné par la lettre grecque γ.
Billet 19.
Cycle de Carnot. Machines de chauffage et de réfrigération. Efficacité du cycle Carnot.
En thermodynamique Cycle Carnot ou Processus Carnot est un processus circulaire réversible composé de deux processus adiabatiques et de deux processus isothermes. Dans le procédé Carnot, le système thermodynamique effectue travail mécanique et échange de la chaleur avec deux réservoirs thermiques qui ont des températures constantes mais différentes. Réservoir avec plus haute température appelé radiateur, et avec une température plus basse - un réfrigérateur.
Le cycle de Carnot doit son nom au scientifique et ingénieur français Sadi Carnot, qui l'a décrit pour la première fois dans son essai « Sur force motrice feu et sur les machines capables de développer cette force" en 1824.
Puisque les processus réversibles ne peuvent se produire qu’à une vitesse infinitésimale, la puissance du moteur thermique dans le cycle de Carnot est nulle. La puissance des moteurs thermiques réels ne peut pas être égale à zéro, donc les processus réels ne peuvent se rapprocher du processus Carnot réversible idéal qu'avec plus ou moins de précision. Dans le cycle de Carnot, un moteur thermique convertit la chaleur en travail avec le rendement le plus élevé possible de tous les moteurs thermiques dont les températures maximale et minimale dans le cycle de fonctionnement coïncident respectivement avec les températures du chauffage et du refroidisseur dans le cycle de Carnot.
Laisser moteur thermique se compose d'un radiateur de température Tn, d'un réfrigérateur de température Tx et Fluide de travail.
Le cycle de Carnot se compose de quatre étapes réversibles, dont deux se déroulent à température constante (isotherme) et deux à entropie constante (adiabatiquement). Il est donc pratique de représenter le cycle de Carnot en coordonnées T (température) Et S (entropie).
1. Expansion isotherme(sur la figure 1 - processus A → B). Au début du processus, le fluide de travail a une température Tn, c'est-à-dire la température du réchauffeur. Le corps est ensuite mis en contact avec un élément chauffant qui lui est transféré de manière isotherme (à température constante). quantité de chaleur Q. Dans le même temps, le volume du fluide de travail augmente, il effectue un travail mécanique et son entropie augmente.
2. Expansion adiabatique(sur la figure 1 - processus B → C). Le fluide de travail est déconnecté du réchauffeur et continue de se dilater sans échange thermique avec l'environnement. Dans ce cas, la température corporelle diminue jusqu'à la température du réfrigérateur Tx, le corps effectue un travail mécanique et l'entropie reste constante.
3. Compression isotherme(sur la figure 1 - processus B → G). Le fluide de travail, ayant une température Tn, est mis en contact avec le réfrigérateur et commence à se comprimer de manière isotherme sous l'action force externe, donnant au réfrigérateur une quantité de chaleur Q. Un travail est effectué sur le corps, son entropie diminue.
4. Compression adiabatique(sur la figure 1 - processus G → A). Le fluide de travail est déconnecté du réfrigérateur et comprimé sous l'influence d'une force externe sans échange thermique avec l'environnement. Dans le même temps, sa température augmente jusqu'à la température du radiateur, un travail est effectué sur le corps, son entropie reste constante.
Cycle de Carnot inversé
DANS thermodynamique des groupes frigorifiques et des pompes à chaleur envisagent cycle de Carnot inversé, composé des étapes suivantes : compression adiabatique due au travail (sur la Fig. 1 - processus B→B) ; compression isotherme avec transfert de chaleur vers un réservoir thermique plus chauffé (sur la Fig. 1 - processus B→A) ; expansion adiabatique (sur la Fig. 1 - processus A→G) ; expansion isotherme avec évacuation de la chaleur d'un réservoir thermique plus froid (sur la Fig. 1 - processus Г→В).


Billet 20.
Deuxième loi de la thermodynamique. Entropie. Troisième loi de la thermodynamique.
Deuxième loi de la thermodynamique- un principe physique qui impose des restrictions sur la direction des processus pouvant se produire dans systèmes thermodynamiques.
La deuxième loi de la thermodynamique interdit ce qu'on appelle machines à mouvement perpétuel du deuxième type, montrant que efficacité ne peut pas être égal à un, car pour un processus circulaire la température du réfrigérateur ne peut pas être égale au zéro absolu (il est impossible de construire un cycle fermé passant par un point de température nulle).
La deuxième loi de la thermodynamique est postulat, non prouvable dans le cadre de la théorie classique thermodynamique. Il a été créé sur la base d'une généralisation de faits expérimentaux et a reçu de nombreuses confirmations expérimentales.
PostulatClausius : "Un processus circulaire est impossible, dont le seul résultat est le transfert de chaleur d'un corps moins chauffé à un autre plus chauffé" (ce processus est appelé Processus Clausius).
PostulatThomas (Kelvin) : « Un processus circulaire est impossible, dont le seul résultat serait la production de travail par refroidissement du réservoir de chaleur »(ce processus est appelé Processus Thomson).
L'entropie d'un système isolé ne peut pas diminuer" (loi de l'entropie non décroissante ).
Cette formulation est basée sur l'idée d'entropie comme fonctions d'état système, qui doit également être postulé.
Dans un état d'entropie maximale, les processus macroscopiques irréversibles (et le processus de transfert de chaleur est toujours irréversible en raison du postulat de Clausius) sont impossibles.
Troisième loi de la thermodynamique (Théorème de Nernst) - principe physique qui détermine le comportement entropieà l'approche températureÀ zéro absolu. Fait partie de postulats thermodynamique, accepté sur la base d'une généralisation d'une quantité importante de données expérimentales.
La troisième loi de la thermodynamique peut être formulée comme suit :
"Augmentation de l'entropie à zéro absolu la température tend vers une limite finie, indépendante de l’état d’équilibre dans lequel se trouve le système..
La troisième loi de la thermodynamique s'applique uniquement aux états d'équilibre.
Puisque, sur la base de la deuxième loi de la thermodynamique, l'entropie ne peut être déterminée que jusqu'à une constante additive arbitraire (c'est-à-dire que ce n'est pas l'entropie elle-même qui est déterminée, mais seulement sa variation). La troisième loi de la thermodynamique peut être utilisée pour déterminer avec précision l’entropie. Dans ce cas, l'entropie du système d'équilibre à température nulle absolue est considérée comme égale à zéro.
La troisième loi de la thermodynamique permet de trouver la valeur absolue de l'entropie, ce qui ne peut se faire dans le cadre de la thermodynamique classique (basée sur les première et deuxième lois de la thermodynamique).
Entropie thermodynamique S, souvent simplement appelé entropie, - quantité physique, utilisé pour décrire système thermodynamique, un des principaux grandeurs thermodynamiques. L'entropie est fonction d'état et est largement utilisé dans thermodynamique, y compris chimique.

