Aldehídos y cetonas. Capítulo iii. protección de grupos funcionales durante la síntesis protección del grupo aldehído en síntesis
Leer también
5.1. Características generales
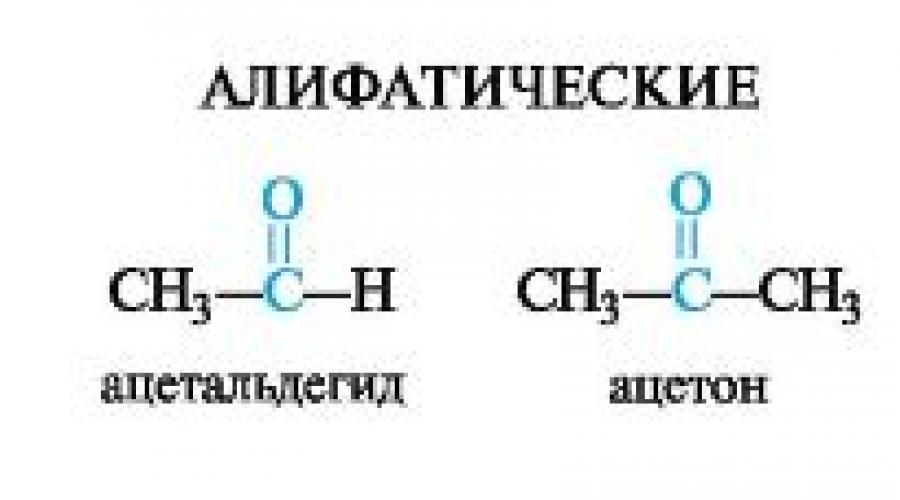
Las clases relacionadas de aldehídos y cetonas contienen un grupo funcional carbonilo y se clasifican como compuestos carbonílicos. También tienen un nombre común. compuestos oxo, ya que el grupo =O se llama grupo oxo.
Los aldehídos son compuestos en los que el grupo carbonilo está unido a un radical orgánico y un átomo de hidrógeno; Las cetonas son compuestos carbonílicos con dos radicales orgánicos.
El grupo -CH=O que forma parte de los aldehídos se llama aldehído, respectivamente, el grupo en cetonas - cetona, o grupo ceto.
Dependiendo de la naturaleza de los radicales orgánicos, los aldehídos y las cetonas pueden pertenecer a alifático o aromático fila; hay cetonas mezclado(Tabla 5.1).

A diferencia de los alcoholes, las moléculas de aldehídos y cetonas no contienen átomos de hidrógeno móviles asociados con átomos de oxígeno. En este sentido, los aldehídos y las cetonas no se unen mediante la formación de enlaces de hidrógeno, pero son propensos a formar enlaces de hidrógeno con moléculas de agua y, por lo tanto, se disuelven bien en ella (especialmente los primeros miembros de la serie homóloga).
Tabla 5.1.Aldehídos y cetonas

5.2. Centros de reacción de aldehídos y cetonas.
sp 2 -El átomo de carbono hibridado del grupo carbonilo forma tres enlaces σ que se encuentran en el mismo plano y un enlace π con el átomo de oxígeno debido al orbital p no hibridado. Debido a la diferencia de electronegatividad de los átomos de carbono y oxígeno, el enlace π entre ellos está muy polarizado (fig. 5.1).
Como resultado, aparece una carga parcial positiva δ+ en el átomo de carbono del grupo carbonilo y una carga parcial negativa δ- en el átomo de oxígeno. Dado que el átomo de carbono tiene deficiencia de electrones, proporciona un sitio para el ataque nucleofílico.

Distribución de la densidad electrónica en moléculas de aldehídos y cetonas, teniendo en cuenta la transferencia de influencia electrónica por electrones.Arroz. 5.1.
Estructura electrónica del grupo carbonilo.
el átomo de carbono deficiente del grupo carbonilo a lo largo de los enlaces σ se presenta en el Esquema 5.1.Centros de reacción en la molécula de aldehídos y cetonas.

Existen varios centros de reacción en las moléculas de aldehídos y cetonas:
El centro electrófilo, el átomo de carbono del grupo carbonilo, determina la posibilidad de un ataque nucleofílico;
El centro principal, el átomo de oxígeno, permite atacar con un protón;
Un centro ácido CH cuyo átomo de hidrógeno tiene una movilidad protónica débil y puede, en particular, ser atacado por una base fuerte.
En general, los aldehídos y las cetonas son muy reactivos.
5.3. Adición nucleofílica
Para los aldehídos y las cetonas, las reacciones de adición nucleofílica son las más características. UN.
Descripción general del mecanismo de adición nucleofílica. UN
La facilidad del ataque nucleofílico al átomo de carbono del grupo carbonilo de un aldehído o cetona depende de la magnitud del ataque parcial.
carga positiva en el átomo de carbono, su accesibilidad espacial y propiedades ácido-base del medio ambiente.

Teniendo en cuenta los efectos electrónicos de los grupos asociados con el átomo de carbono carbonilo, el valor de la carga positiva parcial δ+ en aldehídos y cetonas disminuye en el siguiente orden:

La accesibilidad espacial del átomo de carbono carbonilo disminuye cuando el hidrógeno es reemplazado por radicales orgánicos más voluminosos, por lo que los aldehídos son más reactivos que las cetonas.

Esquema general de reacciones de adición nucleofílica. UN a un grupo carbonilo implica un ataque nucleofílico al átomo de carbono del carbonilo, seguido de la adición de un electrófilo al átomo de oxígeno.

En un ambiente ácido, la actividad del grupo carbonilo generalmente aumenta porque la protonación del átomo de oxígeno crea una carga positiva en el átomo de carbono. La catálisis ácida se suele utilizar cuando el nucleófilo atacante tiene baja actividad.
El mecanismo anterior lleva a cabo una serie de reacciones importantes de aldehídos y cetonas.
Muchas reacciones características de los aldehídos y las cetonas ocurren en condiciones corporales; estas reacciones se presentan en las secciones siguientes del libro de texto. Este capítulo considerará las reacciones más importantes de aldehídos y cetonas, que se resumen en el esquema 5.2.

Adición de alcoholes. Los alcoholes, al interactuar con los aldehídos, se forman fácilmente. hemiacetales. Los hemiacetales no suelen aislarse debido a su inestabilidad. Cuando hay un exceso de alcohol en un ambiente ácido, los hemiacetales se transforman en acetales.

El uso de un catalizador ácido en la conversión de hemiacetal en acetal queda claro a partir del mecanismo de reacción que se indica a continuación. El lugar central en él lo ocupa la formación de un carbocatión (I), estabilizado gracias a la participación de un par solitario de electrones de un átomo de oxígeno vecino (efecto +M del grupo C 2 H 5 O).

Las reacciones de formación de hemiacetales y acetales son reversibles, por lo que los acetales y hemiacetales se hidrolizan fácilmente con exceso de agua en un ambiente ácido. Los hemiacetales son estables en un ambiente alcalino, ya que la alcoxidiona es un grupo saliente más difícil que el ion hidróxido.
La formación de acetales se utiliza a menudo como protección temporal del grupo aldehído.
Conexión de agua. Adición de agua a un grupo carbonilo - hidratación- reacción reversible. El grado de hidratación de un aldehído o cetona en una solución acuosa depende de la estructura del sustrato.
El producto de hidratación, por regla general, no se puede aislar en forma libre mediante destilación, ya que se descompone en sus componentes originales. El formaldehído en una solución acuosa está hidratado en más del 99,9%, el acetaldehído está aproximadamente a la mitad y la acetona prácticamente no está hidratada.

El formaldehído (formaldehído) tiene la capacidad de coagular proteínas. Su solución acuosa al 40%, llamada formalina, utilizado en medicina como desinfectante y conservante de preparaciones anatómicas.
El aldehído tricloroacético (cloral) está completamente hidratado. El grupo triclorometilo, aceptor de electrones, estabiliza tanto el hidrato de cloral que esta sustancia cristalina se separa del agua sólo durante la destilación en presencia de sustancias deshidratantes: ácido sulfúrico, etc.

La base del efecto farmacológico del hidrato de cloral CC1. CH(OH)2 radica el efecto específico sobre el organismo del grupo aldehído, que determina las propiedades desinfectantes. Los átomos de halógeno potencian su efecto y la hidratación del grupo carbonilo reduce la toxicidad de la sustancia en su conjunto.
Adición de aminas y sus derivados. Aminas y otros compuestos que contienen nitrógeno de fórmula general NH. 2 X (X = R, NHR) reacciona con aldehídos y cetonas en dos etapas. En primer lugar, se forman productos de adición nucleófilos que luego, debido a la inestabilidad, eliminan el agua. En este sentido, este proceso generalmente se clasifica como una reacción. eliminación del apego.

En el caso de aminas primarias, sustituidas iminas(también se les llama bases de Schiff).
Las iminas son productos intermedios de muchos procesos enzimáticos. La producción de iminas pasa por la etapa de formación de aminoalcoholes, que son relativamente estables, por ejemplo, cuando el formaldehído reacciona con α-aminoácidos (ver 12.1.4).
Las iminas son productos intermedios en la preparación de aminas a partir de aldehídos y cetonas mediante aminación reductora. Este método general implica reducir una mezcla de un compuesto carbonílico con amoníaco (o una amina). El proceso se desarrolla según el esquema de adición-eliminación con la formación de una imina, que luego se reduce a una amina.
Cuando los aldehídos y las cetonas reaccionan con derivados de hidrazina, producen hidrazonas. Esta reacción se puede utilizar para aislar aldehídos y cetonas de mezclas e identificarlos cromatográficamente.

Las bases de Schiff y otros compuestos similares se hidrolizan fácilmente mediante soluciones acuosas de ácidos minerales para formar los productos de partida.
En la mayoría de los casos, las reacciones de aldehídos y cetonas con bases nitrogenadas requieren catálisis ácida, que acelera la deshidratación del producto de adición. Sin embargo, si la acidez del medio aumenta demasiado, la reacción se ralentizará como resultado de la conversión de la base nitrogenada en el ácido conjugado no reactivo XNH. 3+.
Reacciones de polimerización. Estas reacciones son características principalmente de los aldehídos. Cuando se calientan con ácidos minerales, los polímeros de aldehído se descomponen en sus productos originales.
La formación de polímeros puede considerarse como el resultado de un ataque nucleofílico del átomo de oxígeno de una molécula de aldehído al átomo de carbono carbonílico de otra molécula. Entonces, cuando el formaldehído reposa, el polímero de formaldehído, el paraform, precipita en forma de un precipitado blanco.
5.4. Reacciones de condensación
La presencia de un centro ácido CH en una molécula de aldehído o cetona da como resultado que los átomos de hidrógeno α de estos compuestos carbonílicos tengan cierta movilidad protónica. Bajo la influencia de bases, dichos protones pueden eliminarse para formar los correspondientes carbaniones. Los carbaniones desempeñan el papel de nucleófilos hacia el sustrato carbonilo. Esto permite realizar reacciones en las que una molécula, como nucleófilo, se une al grupo carbonilo de otra molécula de un compuesto carbonílico neutro. Estos procesos pertenecen a reacciones de condensación.
La condensación es una reacción que conduce a la formación de un nuevo enlace carbono-carbono, y se forma una molécula nueva y más compleja a partir de dos o más moléculas relativamente simples.
Así, en un ambiente alcalino, dos moléculas de acetaldehído forman hidroxialdehído con el doble de átomos de carbono.
El producto de reacción que contiene grupos hidroxilo y aldehído se llama aldolem(de palabras aldo egido y alcohol viejo), y la reacción en sí se llamó condensación aldólica, o adición de aldol.
El mecanismo de condensación aldólica. Bajo la acción de una base en un compuesto carbonílico, se elimina un protón de la posición α y se forma un carbonión (I), en el que la carga negativa se deslocaliza con la participación del grupo carbonilo.

El anión (I) es un nucleófilo fuerte (que se muestra en color en el siguiente paso del mecanismo) que se suma a la segunda molécula (no ionizada) del compuesto carbonílico. Como resultado de esta interacción, aparece un nuevo enlace C-C y se forma un ion alcóxido intermedio (II). En un ambiente acuoso, este anión se estabiliza eliminando un protón de una molécula de agua y se convierte en el producto final: un aldol.

La reacción de adición de aldol se muestra usando el ejemplo de propanal (la molécula que se une al grupo C=O de otra molécula está resaltada en color); Se muestra una reacción similar usando acetona como ejemplo.

El producto de condensación, el aldol, es capaz de eliminar agua para formar un compuesto carbonílico α,β-insaturado. Esto suele ocurrir a temperaturas elevadas. En este caso, la reacción en su conjunto se llama Condensación de crotón.
Las reacciones de condensación también pueden ocurrir en una versión mixta, utilizando diferentes compuestos carbonílicos, y uno de ellos puede no contener un centro ácido CH, como el formaldehído y el benzaldehído en las siguientes reacciones:

La condensación aldólica es una reacción reversible; el proceso inverso se llama escisión aldólica(o reacción de retroaldol). Ambas reacciones ocurren en muchos procesos bioquímicos.
5.5. Reducción y oxidación.
Recuperaciónlos aldehídos y cetonas se llevan a cabo utilizando hidruros metálicos complejos LiAlH 4, NaBH 4. La reacción implica un ataque nucleofílico al átomo de carbono carbonilo por un ion hidruro.
La hidrólisis posterior del alcoholato resultante produce un alcohol primario o secundario.

OxidaciónLa transformación de aldehídos en ácidos carboxílicos se lleva a cabo bajo la influencia de la mayoría de agentes oxidantes, incluido el oxígeno atmosférico. Las cetonas no se oxidan en condiciones suaves.

El óxido de plata en forma de complejo de amoníaco 2 OH (reactivo de Tollens) oxida los aldehídos en ácidos carboxílicos, liberando plata metálica. De ahí viene el nombre: reacción. "espejo plateado"
Los aldehídos también se oxidan fácilmente con hidróxido de cobre (II) en un medio alcalino.
Ambas reacciones se utilizan a menudo cualitativamente para detectar el grupo aldehído, aunque no son específicas con respecto a los aldehídos: por ejemplo, los fenoles polihídricos, aminofenoles, aminas aromáticas, hidroxicetonas y otros compuestos fácilmente oxidados están sujetos a oxidación por estos reactivos.
R-C-OR" + ROH: N
En la hidrólisis alcalina, el grupo saliente (RO®) es | Parece muy malo y no es posible reaccionar. Esta propiedad, la estabilidad de los acetales en un ambiente alcalino, se utiliza cuando es necesario proteger el grupo carbonilo. La protección de uno u otro grupo funcional (en aminas, alcoholes, fenoles, olefinas, mercaptanos, ácidos CH, etc.) es una tarea muy importante en la síntesis orgánica (Capítulo XXII [lo ilustramos con el ejemplo de la síntesis de). aldehído de glicerol a partir de acroleína fácilmente disponible.
CH2=CH-Cf° + KMnO, N
La acción del permanganato de potasio directamente sobre la acroleína conduce a la oxidación tanto de ^C=CX^ como del grupo aldehído-C: CH2-CH-C je je je
acroleína ácido glicérico uO HC1.
Esto requiere la protección del grupo aldehído, lo que se puede lograr convirtiéndolo en acetal, por ejemplo, mediante la acción del etanol en presencia de cloruro de hidrógeno.
3-cloropropanal
CH2-CH2-C-OS2H5
1D-dietoxi-3-xiaorpropano
Este último se une inmediatamente al doble enlace simultáneamente con la formación de un acetal. La etapa clave de la síntesis es la regeneración del doble enlace C=C como resultado de la decloración con álcali con la preservación de un acetal que es estable en un ambiente alcalino.
CH2-CH-Cr-OC2H5
OH OH H 1, 1-dietoxi-2,3-dihidroxipropano
La hidrólisis ácida del acetal en condiciones suaves da el gliceraldehído deseado:
R°g^ H3Oe UR
sn2-sn-schn? sn2-sn-on él os2n5 él él n
2,3-dihidroxipropanal, gliceraldehído Debido al impedimento estérico, las cetonas reaccionan con los alcoholes para formar hemicetales mucho más difícilmente en comparación con los aldehídos que forman hemiacetales, especialmente con grupos voluminosos en la cetona o el alcohol.
Para proteger el grupo carbonilo es conveniente utilizar glicoles que formen acetales cíclicos, por ejemplo:
^O c© ^o-sn2
CH3CH2CH- + CH2-CH2 -H- CH3-CH2-C I
N OH OH N 0 СНз
2-ETHL-1,3-dioxalano
Esto es importante principalmente para las cetonas, que no tienden a formar cetales cuando interactúan con alcoholes comunes. La formación intramolecular de hemiacetales por oxialdehídos y oxicetonas es característica de los carbohidratos, véase el Capítulo XXIII para más detalles.
Adición de ácidos carboxílicos. A los aldehídos, por analogía con los alcoholes, se les pueden añadir ácidos carboxílicos (preferiblemente sus anhídridos), formando acilos:
acetaldehído anhídrido acético diacetato de etilideno
Polimerización de aldehídos. Los aldehídos inferiores (formaldehído, peor aún, acetaldehído) son capaces de polierizarse, cuyo iniciador suele ser el agua.
Nosn2-o-sn2-on + n-s
Etc. - HO^CH2O^H
¿La naturaleza de los productos poliméricos depende de las condiciones?
W En soluciones acuosas, el formaldehído forma polímeros lineales oligoméricos. Cuando se evapora dicha solución, se forma un producto sólido, el paraformaldehído. que contiene de 8 a 100 unidades de oximetileno. El agua, al iniciar la polimerización, descompone simultáneamente el polímero, hidrolizándolo, por lo que es imposible obtener un polímero de alto peso molecular en soluciones acuosas cuando se calienta, el formaldehído de ratán, especialmente con ácidos, se descompone y se convierte en formaldehído gaseoso, |^si; esto sucede en un recipiente cerrado - en trioxano "."pl. 64 "C, punto de ebullición 115°C).
trioxano
pero-|-сн2о--н -?- 9^у
paraformaldehído
La tentadora idea de producir un polímero de alto peso molecular (L > 1000) a partir de formaldehído atrajo a muchos químicos famosos. El poliformaldehído fue descrito por primera vez por A. M. Butlerov a mediados del siglo XIX. El polímero renació gracias al trabajo del químico alemán G. Staudinger, uno de los fundadores de la química de polímeros, quien llevó a cabo una investigación fundamental básica sobre la síntesis y las propiedades del poliformaldehído de alto peso molecular, incluidos métodos químicos para aumentar su estabilidad. Sin embargo, fue posible superar las enormes dificultades en la implementación técnica de la síntesis y establecer la producción y el procesamiento industrial del poliformaldehído de alto peso molecular por primera vez en 1959 (Dupont).
Actualmente, el poliformaldehído se obtiene en forma de homopolímero con grupos hidroxi terminales convertidos en simples para evitar la despolimerización.
o ésteres (Delrin, Tenac), o copolímero de formaldehído con 2,5-3,0% de óxido de etileno, 1,3-dioxolano
(I J) y otros (celcon, SFD, hostaform) con O molecular
con un peso de 40 a 120 mil.
CH3-C-O-J-CH2OJ-C-CH3
poliformaldehído (delrin, tenac)
El poliformaldehído, como excelente material de construcción, se utiliza cada vez más en maquinaria, fabricación de instrumentos y para hilar fibras.
79.3.1.3. Reacciones con nucleófilos centrados en halógenos.
Los halogenoaniones son nucleófilos débiles (buenos grupos salientes) y el HHal forma productos de adición inestables con aldehídos y cetonas, como se señaló anteriormente.
En todas las aproximaciones al problema de la selectividad que hemos considerado anteriormente, el “juego” se basaba en variaciones que afectaban directamente a los participantes en el proceso principal: la naturaleza del sustrato y/o reactivo, las condiciones de reacción o incluso la naturaleza. de la reacción misma cambió. Si bien en cada caso fue posible asegurar la selectividad de la transformación requerida, a veces este éxito se logró a un alto precio, ya que fue necesario “ajustar” cualquiera de los principales métodos de síntesis a la solución de un problema particular, en otros palabras, utilizando la metáfora utilizada anteriormente de "meterse dentro de una caja negra". En la práctica, en muchos casos . Resulta más ventajoso un enfoque diferente del problema de la selectividad. Expliquemoslo con el siguiente ejemplo esquemático.
Consideremos un determinado sustrato A-X, para el cual se ha desarrollado bien el método para convertirlo en el producto A-Z. Supongamos ahora que la tarea específica es la conversión selectiva del sustrato Y-A-X, donde Z es un grupo similar en propiedades al grupo X, en el producto Y-A-Z. Por supuesto, puede intentar, por ejemplo, modificar la reacción principal para que afecte sólo al grupo X y no afecte en absoluto al grupo Y. Sin embargo, este camino puede resultar muy laborioso, ya que lo hará. Será necesario modificar un método ya bien desarrollado y posiblemente complejo, y es posible que para cada nuevo Y en sistemas del tipo Y "-A-X este trabajo deba realizarse de nuevo. Afortunadamente, existe un principio diferente para resolver este problema. tipo de problema La esencia del ggo es eliminar temporalmente el grupo Y del juego y así transformar el sustrato bifuncional Y-A-X en uno monofuncional, al que se puede aplicar el método habitual de transformar X en Z en su forma canónica. se puede lograr mediante el uso de algunas reacciones simples que transforman la función Y en un grupo que es inerte en las condiciones de la reacción principal y permite un retorno indoloro de la función Y original en etapas posteriores de síntesis.
Este enmascaramiento, o protección de funciones, es una técnica muy utilizada en la práctica de la síntesis orgánica. Es fácil ver que esto elimina el problema de la selectividad de la reacción principal, pero surge la pregunta sobre la selectividad de colocar un grupo protector en la función Z sin afectar la función relacionada X. Sin embargo, en el caso general, encontrar una solución solucionar este problema ya es incomparablemente más fácil por varias razones. En primer lugar, los métodos para introducir la protección pertenecen a la categoría de transformaciones de grupos funcionales, que son relativamente simples en química y para las cuales se han desarrollado decenas de métodos, lo que los hace aplicables a casi todos los casos imaginables. En segundo lugar, la estructura del grupo protector se puede variar dentro de límites muy amplios, ya que se eliminará en etapas posteriores y su naturaleza no puede afectar la formación de productos posteriores de la cadena sintética*. Debido a estas circunstancias, la gama de reacciones que se pueden utilizar para proteger un grupo funcional determinado es extremadamente amplia, lo que garantiza de forma fiable la selectividad necesaria del grupo protector. Para ilustrar la aplicación del “enfoque protector” al problema de la selectividad, consideremos la restauración del ya conocido modelo de sistema trifuncional 156 (Diagrama 2.86).
| Esquema 2.86 |
Anteriormente, usando el mismo sistema, mostramos cómo se puede lograr la reducción selectiva solo del grupo formilo o de los grupos formil icarbometoxi variando la naturaleza del agente reductor hidruro (ver Esquema 2.73). Pero, ¿qué pasa si desea restaurar selectivamente sólo el grupo carbometano? Teniendo en cuenta que esta función será menos activa con respecto a cualquiera de los agentes reductores de hidruro convencionales que el grupo formilo, puede parecer que la transformación requerida no se puede llevar a cabo en absoluto utilizando reactivos de este tipo. Sin embargo, de hecho, la situación se puede corregir fácilmente protegiendo el grupo carbonilo convirtiéndolo en un grupo acetal utilizando, por ejemplo, una reacción catalizada por ácido con etilenglicol. Dado que los acetales son estables para una amplia variedad de nucleófilos, el grupo éster del sustrato modificado 188 se puede reducir usando cualquier agente reductor de hidruro. El alcohol resultante 189 difiere del producto requerido 190 sólo en presencia de protección acetilo, pero este último se elimina fácilmente mediante hidrólisis catalizada por ácido. Por tanto, el problema casi intratable de la reducción selectiva del grupo carbometoxi en presencia de una función aldehído fácilmente reducible se resuelve fácilmente utilizando un "enfoque protector".
Veamos ahora más específicamente algunos métodos para proteger los grupos funcionales más importantes, comenzando con la función carbonilo.
La protección total mencionada anteriormente se puede aplicar, en principio, a cualquier compuesto carbonílico utilizando una amplia variedad de alcoholes o glicoles, pero la velocidad de esta reacción, dependiendo de la naturaleza específica del sustrato, puede variar en varios órdenes de magnitud. Esto permite, en particular, diferenciar claramente las funciones aldehído y cetona, ya que la primera es un electrófilo más activo y puede convertirse mucho más fácilmente en acetal. Consideremos como ejemplo un problema sintético específico en el que se utilizó eficazmente esta técnica particular.
Usando el mismo ejemplo, es conveniente mostrar cómo se puede asegurar la selectividad inversa de la reducción. Para ello, primero se protege el grupo aldehído mediante protección tioacetal (Esquema 2.88). Dado que los tioacetales son bastante estables en condiciones ligeramente ácidas, el producto resultante 194 se puede convertir aún más en un derivado desprotegido 195. Una característica específica de los tioacetales es su capacidad de sufrir solvólisis con bastante facilidad cuando se tratan con sales de mercurio (o cadmio). Mediante dicho procesamiento del producto. 195 obtener un derivado monosustituido 196, en el que esta vez el grupo ceto está protegido y el grupo aldehído puede reducirse aún más o usarse en cualquier otra reacción con reactivos nucleofílicos.
A menudo hay casos en los que es necesario diferenciar entre un grupo carbonilo regular y el mismo grupo conjugado con un doble enlace. Dado que la presencia de dicha conjugación reduce significativamente la electrofilia del centro carbonilo, la acetalización en tales sistemas polifuncionales se producirá con una alta selectividad, afectando sólo a la función carbonilo aislada. Esta técnica, que se utiliza especialmente en la química de los esteroides, permite en etapas posteriores utilizar el grupo enona conservado en la molécula en transformaciones como, por ejemplo, la adición de Michael.
Conviene considerar los problemas que surgen cuando es necesario realizar una protección selectiva de los grupos hidroxilo utilizando ejemplos de la química de los carbohidratos. Digamos que necesitamos llevar a cabo selectivamente la reacción en el grupo hidroxilo primario en C-6 del a-metil-O-glucopiranósido. (197) (diagrama 2.89).
Evidentemente, para lograr este objetivo es necesario proteger primero las otras tres funciones hidroxilo presentes en la molécula. Una posible forma de solucionar este problema es la síntesis de triacetato. 198. Sin embargo, la transformación directa 197 V 198 difícil de realizar, porque la acetilación es una reacción poco selectiva que ocurre más rápido con los alcoholes primarios que con los secundarios. Por lo tanto, tenemos que recurrir a una solución alternativa: la síntesis de éter trifenilmetilo (tritilo, Tg). 199. La introducción de protección tritilo en los hidroxilos primarios es más fácil que en los secundarios, ya que las reacciones del grupo tritilo voluminoso son muy sensibles al blindaje espacial del centro atacado. De hecho, el tratamiento con glucósidos 197 El cloruro de tritilo en piridina conduce a éter monotritilo con alto rendimiento. 199. En este compuesto, el hidroxilo primario está protegido, que debería estar libre en el compuesto objetivo. Esto, sin embargo, no debería confundirnos: lo principal es que logramos “etiquetarlo” de alguna manera, es decir. distinguir de los demás. En la siguiente etapa, debemos cerrar todos los demás grupos hidroxilo, para lo cual es muy posible utilizar el método estándar de acetilación con anhídrido acético en piridina. En la derivada resultante 200 Hay dos tipos de grupos protectores, que se diferencian marcadamente en sus propiedades, en particular en su estabilidad frente a reactivos ácidos. Por lo tanto, convertir este producto al triacetato objetivo 198 Puede llevarse a cabo con alta selectividad mediante hidrólisis en un ambiente ligeramente ácido.
| Esquema 2.89 |
Utilizando el ejemplo considerado, resulta instructivo trazar algunos principios generales para el uso de grupos protectores. La selectividad del resultado final en la secuencia de transformaciones mostrada se consigue, por un lado, por la selectividad de introducir la primera protección, tanto por sus propiedades como por las propiedades de la función protegida, y por otro lado, por la selectividad de eliminar una de las protecciones, debido únicamente a diferencias en las propiedades de estos grupos como tales. Por lo tanto, la selectividad de la introducción de protección y la selectividad de la desprotección están controladas por factores completamente diferentes y, por lo tanto, constituyen dos formas poderosas e independientes de controlar la selectividad de toda la síntesis.
El problema de la protección selectiva del grupo hidroxilo surge muy a menudo en la síntesis total. Por eso se ha creado un sistema de protección muy sofisticado para la función del alcohol, literalmente “para todas las ocasiones”. Algunas de las protecciones más utilizadas se muestran en el Diagrama 2.90. Todos los derivados mostrados se encuentran entre los productos de transformación del grupo hidroxilo generalmente bastante comunes: estos son ésteres (201-203), acetales (204, 205), éteres (206-209) y éteres sililicos (210, 211) . La preparación de todos estos derivados se realiza según el esquema general de sustitución electrófila del hidrógeno del grupo hidroxilo, sin embargo, los métodos para introducir protecciones específicas varían mucho y abarcan tanto la región ácida, como la neutra y la alcalina. La facilidad de la reacción para establecer una protección particular depende de la naturaleza del alcohol hidroxilo, es decir, de las características estructurales del fragmento que contiene el sustituyente hidroxilo. Así, por ejemplo, la reactividad relativa de los alcoholes en tales reacciones se puede representar mediante la serie: “-AlkOH > v/ao/>-A1UN > terc-MkOI; ROH ecuatorial > ROH axial. Aprovechando las diferencias en la reactividad de las funciones del alcohol, es posible diferenciar estos grupos de forma bastante sutil mediante la introducción selectiva de protecciones adecuadas.
El rango de condiciones bajo las cuales la protección de los hidroxilos de alcohol es estable cubre casi toda la región en la que se pueden llevar a cabo las principales reacciones utilizadas en la síntesis orgánica (excepto los medios superácidos). En general, los éteres, acetales y cetales se caracterizan por una alta resistencia a bases y nucleófilos, así como a agentes oxidantes y reductores; para ésteres, a electrófilos y agentes oxidantes y, en una gama bastante amplia, a ácidos; para éteres sililicos, hasta agentes oxidantes y reductores y electrófilos de algunos tipos. Por lo tanto, para garantizar la seguridad del grupo de los alcoholes en condiciones de casi cualquier reacción que ocurra con la participación de otras funciones disponibles, siempre es posible seleccionar algún tipo de protección entre el rico conjunto de opciones disponibles.
| Esquema 2.90 |
Las condiciones para eliminar las protecciones enumeradas también son muy diversas: son solvólisis ácida o alcalina, hidrogenólisis catalítica, reducción con hidruros complejos o metales alcalinos en amoníaco líquido y escisión bajo la influencia de reactivos específicos como, por ejemplo, ion fluoruro no solvatado ( para derivados de sililo) o trimetilyodosilano (para ésteres metílicos, estable a la mayoría de los demás reactivos). Dentro de cada tipo de protección existen sutiles gradaciones de resistencia en relación con las condiciones en las que se eliminan. Por ejemplo, en el grupo de los ésteres, la resistencia a la solvólisis alcalina aumenta en la serie: ChCCOO-R.< C1CH 2 COO-R < CH 3 COO-R < C 6 H 5 COO-R < QHsNHCOO-R. Аналогично изменяется стабильность силиловых эфиров в условиях сольволиза в ряду: Me 3 Si-O-R < Me 3 CSi(Me 2)-О-R < МезС81(Рп 2)-О-R. Очень важной является возможность удаления силиль-ной группы при действии фторид-иона, что позволяет снимать эту группу, не затрагивая какие-либо другие защиты. В группе простых эфиров резко различными будут условия снятия защит при замене алкильной группы на ал-лильную, бензильную или тритильную. Так, удобным методом снятия ал-лильной защиты является двустадийная процедура: изомеризация в пропе-ниловый эфир под действием /я/>e/r-butilag potásico en DMSO absoluto (o bajo la acción de complejos de rodio) e hidrólisis en condiciones ligeramente ácidas (ver Esquema 2.90). El grupo bencilo se puede eliminar en condiciones neutras mediante hidrogenólisis sobre un catalizador de paladio o mediante reducción de un electrón con sodio en amoníaco líquido. Tritap y su análogo cercano de protección p-metoxitritilo son muy similares en sus propiedades, pero difieren tanto en la velocidad de solvólisis ácida que eliminar el grupo p-metoxitritilo mientras se preserva el grupo tritilo no es un problema particular.
La variedad de métodos para proteger la función hidroxilo, así como métodos para eliminar grupos protectores, es una poderosa herramienta que facilita enormemente la solución de todo tipo de problemas sintéticos, de una forma u otra relacionados con el uso de funciones alcohólicas. Entre ellos puede haber no sólo tareas asociadas con la producción selectiva de ciertos derivados en una serie de compuestos polihidroxilados, como, por ejemplo, se muestra en el Esquema 2.89. En una síntesis completa, es muy importante utilizar un sistema de protección configurado de tal manera que permita utilizar un precursor polifuncional como sustrato en una secuencia de transformaciones controladas que afectan a estas funciones una tras otra.
Un claro ejemplo del éxito de este enfoque -un enfoque que es estratégico en su significado- es la síntesis del diterpenoide natural biológicamente activo zoopathenol (212), llevada a cabo por Nikolaou et al. . El análisis retrosintético de esta estructura sugirió un desmontaje en los enlaces a, byc, lo que permitió seleccionar la bromcetona 213 y el triol 214 como los principales bloques sintéticos (Esquema 2.91). La ruta formal para la síntesis del producto objetivo a partir de estos iniciales, incluida una secuencia de varias transformaciones, también se muestra en el Esquema 2.91 (los asteriscos indican aquellos centros en los reactivos que participan en la formación de enlaces en cada etapa).
Desde el punto de vista de la estrategia general, este plan parece bastante convincente, ya que implica relativamente pocas etapas, cada una de las cuales implica el uso de reacciones bien conocidas. Sin embargo, incluso con un análisis superficial, queda claro que es simplemente imposible implementarlo en la forma presentada debido a obstáculos prácticamente insuperables causados por la naturaleza multifuncional de todos los reactivos 213-218 mostrados en esta secuencia hipotética. Así, por ejemplo, aunque técnicamente es posible imaginar la formación de un enlace C-C cuando 215 se ensambla a partir de los precursores 213 y 214 según el esquema de reacción de Grignard entre el aldehído obtenido por oxidación de 214 y el compuesto organomagnesio preparado a partir del bromuro 213. , es imposible oxidar directamente 214 al aldehído de la estructura requerida, así como obtener el reactivo de Grignard a partir de 213 (debido a la presencia de un electrófilo carbonilo en esta molécula). Es fácil ver que la implementación de otras etapas de la secuencia mostrada es igualmente imposible en la realidad, a pesar de la presencia de métodos bien desarrollados para llevar a cabo estas transformaciones.
| Esquema 2.91 |
Evidentemente, sería absolutamente inútil intentar implementar al menos una de las etapas de este plan con los sustratos 213-218. Sin embargo, de hecho, la síntesis de 212 se llevó a cabo con éxito en total conformidad con el plan mostrado anteriormente y utilizando los compuestos 213 y 214 como materiales de partida, que, sin embargo, se incluyeron en la cadena sintética en forma de derivados protegidos (ver Esquema 2.92).
El equivalente sintético del triol 214 fue el derivado 219, en el que los tres grupos hidroxi están protegidos de manera diferente. La eliminación selectiva de la protección del tetrahidropiranilo libera el hidroxilo primario deseado, que se oxida aún más al aldehído 220 deseado. Como se señaló, el cetobromuro 213 no se puede usar directamente para preparar el reactivo de Grignard correspondiente. Sin embargo, no hay nada que impida la conversión de 213 al cetal correspondiente, a partir del cual se puede obtener fácilmente el reactivo requerido 221. La reacción de 220 con 221, la posterior oxidación del único grupo hidroxilo desprotegido del producto 222 y una repetición de Grignard. La reacción sobre el grupo carbonilo resultante no presenta ningún problema. El producto 223 contiene dos dobles enlaces, pero sólo uno de ellos debe convertirse en el epóxido necesario para la posterior construcción del anillo de oxepano. Para la epoxidación de 223, no se pueden utilizar los reactivos más utilizados para este fin, como los perácidos, porque atacarán principalmente el doble enlace trisustituido más nucleofílico. Para proporcionar la selectividad de oxidación requerida, se eliminó la protección de sililo (mediante la acción de un anión flúor no solvatado) y el alcohol alílico resultante se oxidó adicionalmente con tert-ВuUN - Reactivo para la epoxidación selectiva de dobles enlaces en alcoholes alílicos. La etapa clave de toda la síntesis, la ciclación intramolecular del epóxido 224 con la formación de un anillo de siete miembros, se desarrolla de forma bastante selectiva, ya que el hidroxilo secundario, el competidor más peligroso del grupo hidroxilo terciario reactivo, está protegido de forma fiable. El producto de ciclación diol 225 se convirtió adicionalmente en cetona 226 mediante oxidación estándar del resto 1,2-diol, después de lo cual sólo fueron necesarias unas pocas transformaciones bastante triviales para completar la síntesis de 212.
| Esquema 2.92 |
Es obvio que el éxito de toda la síntesis estuvo determinado principalmente por una elección cuidadosamente pensada del sistema de grupos protectores en los compuestos de partida. De hecho, la presencia de tres grupos protectores diferentes en 219, un derivado del triol de partida 214, permitió eliminar cada uno de ellos precisamente en el momento en que era necesario realizar una u otra transformación que implicara selectivamente una función hidroxilo específica. y colocar protección en la función cetona en el bromuro 213 garantizó la seguridad del fragmento cetónico en toda la secuencia sintética. Es de destacar que en la síntesis de esta estructura objetivo multifuncional, se minimizaron las manipulaciones con grupos protectores y no fue necesaria la inclusión de operaciones auxiliares para configurar y eliminar protecciones adicionales en ninguna etapa.
Hasta ahora hemos hablado de compuestos protegidos como derivados que aseguran la conservación de una función particular en condiciones de transformaciones sintéticas. Sin embargo, muchas veces un mismo grupo puede servir como protector en una serie de reacciones y funcional en otra. A continuación se discutirán algunos ejemplos que ilustran la importancia de este aspecto del uso de grupos protectores en síntesis.
Quizás el más simple y obvio sea el caso de la protección de éster del grupo de los alcoholes. Como señalamos anteriormente, esta protección permite preservar la función del alcohol en condiciones de reacciones como la oxidación o la glicosilación. Sin embargo, no menos importante desde el punto de vista sintético es la capacidad de los ésteres, especialmente los trifluoroacetatos o triflatos, de servir como electrófilos activos en reacciones con nucleófilos carbaniónicos para formar un enlace C-C (véase, por ejemplo, el esquema 2.79).
Otra forma clásica de proteger los alcoholes es convertirlos en éteres tritílicos. Muy a menudo, este método se utiliza para excluir la posibilidad de sustitución electrófila de hidrógeno en el grupo hidroxilo correspondiente. Sin embargo, en el caso de los alcoholes secundarios, la transición a grupos tritilo facilita significativamente la extracción del ion hidruro del fragmento a-CH bajo la acción de catalizadores específicos como el catión tritilo, por lo que puede ocurrir fácilmente una desproporción. con la formación de un fragmento de cetona y trifenilmetano. El esquema 2.93 muestra un ejemplo del uso de esta característica de protección tritilo para llevar a cabo la oxidación selectiva de un grupo alcohol secundario en un sustrato bifuncional 227 .
| Esquema 2.93 |
Es bien sabido que la transformación de un aldehído carbonilo en una función ditioacetal garantiza la seguridad de este carbonilo en condiciones de reacciones de adición nucleofílica, oxidación o reducción de hidruro. Pero no menos importante para la síntesis es el hecho de que los ditioacetales pueden servir como precursores convenientes para la generación de los reactivos carbaniones correspondientes (bajo la acción de bases como el butillitio), y en la siguiente sección analizaremos más de cerca los detalles de esta aplicación de ditioacetales.
La conversión de cetonas en cetales es un método tradicional para proteger este fragmento en condiciones de reducción, especialmente útil en los casos en los que es posible una protección selectiva en uno de los grupos carbonilo del sustrato. Por lo tanto, el monocetal 228 (Esquema 2.94) se puede obtener fácil y selectivamente a partir de la dicetona correspondiente, ya que el segundo grupo cetona (en C-17) en este compuesto está estéricamente impedido. La reducción de 228 con borohidruro de sodio da (después de la hidrólisis del grupo protector) el cetoalcohol 229 con un rendimiento casi cuantitativo; el resultado se puede decir que es banal. Sin embargo, resulta que al reducir el mismo sustrato 228, se puede asegurar la regioselectividad inversa con la misma integridad, es decir, reducción exclusiva en el centro C-3. Este resultado, a primera vista paradójico, se consigue si la reducción se lleva a cabo con diyodosilano, un reactivo para la escisión específica y la hidrogenólisis del grupo dioxolano. Así, en la reacción 228 → 230, el grupo cetal (¡sólo un equivalente disfrazado del grupo ceto!) actúa como una función con propiedades bastante inusuales.
| Esquema 2.94 |
Entre los derivados ácidos, las amidas ocupan un lugar especial debido a su electrofilia reducida y, en consecuencia, a su mayor estabilidad en las condiciones de los métodos utilizados habitualmente para descomponer otros derivados carboxílicos. Sin embargo, en general, la protección de amida no se utiliza muy a menudo en síntesis precisamente debido a las estrictas condiciones necesarias para la regeneración de la función carboxilo (ver ejemplos en el trabajo). Sin embargo, fue con el uso de amidas que fue posible simplificar significativamente la solución de los problemas de selectividad en la reacción de Michael en la serie de derivados de ácidos a,p-insaturados. Así, se sabe que la interacción de ésteres de tales ácidos con compuestos organomagnesicos o de litio conduce habitualmente a la formación de mezclas de productos de adición 1,2 y 1,4. En algunos casos (¡pero no en todos!), el problema de la producción selectiva de aductos 1,4 se puede resolver utilizando reactivos de cuprato. La situación se simplifica enormemente si tomamos dimetilamidas como 231 (ver diagrama 2.95) como aceptadores de Michael. Debido a la presencia del fragmento de dimetilamida, el ataque del nucleófilo al átomo de carbono del carbonilo está completamente bloqueado y las reacciones con reactivos de organolitio de diversas naturalezas se desarrollan exclusivamente como una adición 1,4. Además, el carbanión intermedio formado en la primera etapa es suficientemente estable en las condiciones de adición de Michael, lo que permite introducirlo en reacciones con una amplia gama de electrófilos y así obtener una variedad de productos de adición de nucleófilos C y electrófilos C. en el doble enlace del tipo de sustrato 231. Se puede lograr el mismo resultado cuando se trabaja con trimetilhidrazidas de ácidos, como 232 .
| Esquema 2,95 |
Esta sección ha esbozado algunos principios generales para el uso de grupos protectores, con ejemplos relacionados con la química del alcohol y, en menor medida, de los grupos carbonilo. Hasta la fecha, se ha desarrollado un sistema muy sofisticado de protección para casi todos los grupos funcionales principales y se continúa investigando intensamente en esta área. Así, en la primera edición de la monografía sobre grupos protectores (Verde, Protective Groups in Chemistry, 1981) describe aproximadamente 500 protecciones diferentes para cinco tipos de grupos funcionales. Cuando se publicó la segunda edición de esta monografía en 1991)