Aldehydes and ketones. Chapter iii. protection of functional groups during synthesis protection of aldehyde group in synthesis
5.1. general characteristics
Related classes of aldehydes and ketones contain a carbonyl functional group and are classified as carbonyl compounds. They also have a common name oxo compounds, since the =O group is called an oxo group.
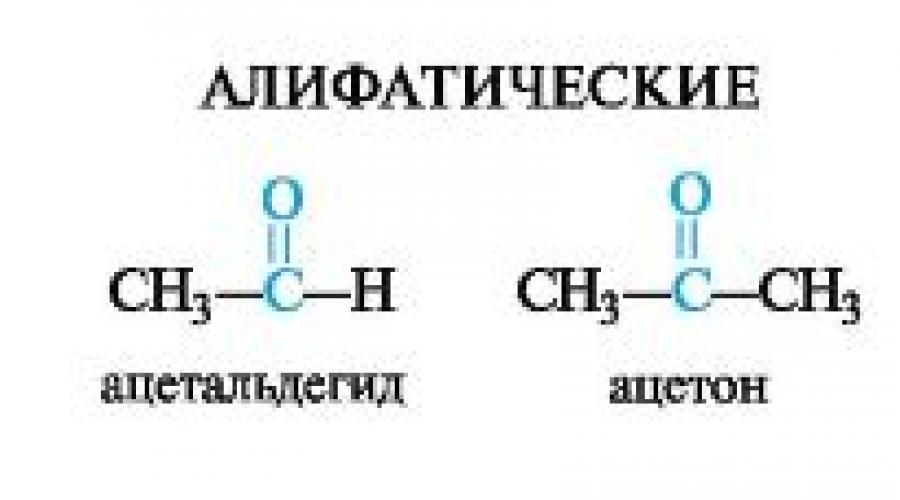
Aldehydes are compounds in which the carbonyl group is bonded to an organic radical and a hydrogen atom; ketones are carbonyl compounds with two organic radicals.
The -CH=O group that is part of aldehydes is called aldehydic, respectively, the group in ketones - ketone, or keto group.
Depending on the nature of organic radicals, aldehydes and ketones may belong to aliphatic or aromatic row; there are ketones mixed(Table 5.1).

Unlike alcohols, the molecules of aldehydes and ketones do not contain mobile hydrogen atoms associated with oxygen atoms. In this regard, aldehydes and ketones are not associated due to the formation of hydrogen bonds, but are prone to form hydrogen bonds with water molecules and therefore dissolve well in it (especially the first members of the homologous series).
Table 5.1.Aldehydes and ketones

5.2. Reaction centers of aldehydes and ketones
sp 2 -The hybridized carbon atom of the carbonyl group forms three σ bonds lying in the same plane and a π bond with the oxygen atom due to the unhybridized p orbital. Due to the difference in electronegativity of carbon and oxygen atoms, the π bond between them is highly polarized (Fig. 5.1). As a result, a partial positive charge δ+ appears on the carbon atom of the carbonyl group, and a partial negative charge δ- appears on the oxygen atom. Since the carbon atom is electron deficient, it provides a site for nucleophilic attack.
Distribution of electron density in molecules of aldehydes and ketones, taking into account the transfer of electronic influence by electron-

Rice. 5.1.Electronic structure of the carbonyl group
the deficient carbon atom of the carbonyl group along σ-bonds is presented in Scheme 5.1.
Scheme 5.1.Reaction centers in the molecule of aldehydes and ketones

There are several reaction centers in the molecules of aldehydes and ketones:
The electrophilic center - the carbon atom of the carbonyl group - determines the possibility of nucleophilic attack;
The main center - the oxygen atom - makes it possible to attack with a proton;
A CH acid center whose hydrogen atom has weak proton mobility and can, in particular, be attacked by a strong base.
In general, aldehydes and ketones are highly reactive.
5.3. Nucleophilic addition
For aldehydes and ketones, nucleophilic addition reactions are most characteristic A N.
General description of the mechanism of nucleophilic addition A N
The ease of nucleophilic attack on the carbon atom of the carbonyl group of an aldehyde or ketone depends on the magnitude of the partial
positive charge on the carbon atom, its spatial accessibility and acid-base properties of the environment.

Taking into account the electronic effects of groups associated with the carbonyl carbon atom, the value of the partial positive charge δ+ on it in aldehydes and ketones decreases in the following order:

The spatial accessibility of the carbonyl carbon atom decreases when hydrogen is replaced by bulkier organic radicals, so aldehydes are more reactive than ketones.

General scheme of nucleophilic addition reactions A N to a carbonyl group involves a nucleophilic attack on the carbonyl carbon atom, followed by the addition of an electrophile to the oxygen atom.

In an acidic environment, the activity of the carbonyl group generally increases because protonation of the oxygen atom creates a positive charge on the carbon atom. Acid catalysis is usually used when the attacking nucleophile has low activity.
A number of important reactions of aldehydes and ketones are carried out by the above mechanism.
Many reactions characteristic of aldehydes and ketones occur under body conditions; these reactions are presented in subsequent sections of the textbook. This chapter will consider the most important reactions of aldehydes and ketones, which are summarized in Scheme 5.2.

Addition of alcohols. Alcohols, when interacting with aldehydes, easily form hemiacetals. Hemiacetals are usually not isolated due to their instability. When there is an excess of alcohol in an acidic environment, hemiacetals transform into acetals.

The use of an acid catalyst in the conversion of hemiacetal to acetal is clear from the reaction mechanism given below. The central place in it is occupied by the formation of a carbocation (I), stabilized due to the participation of a lone pair of electrons of a neighboring oxygen atom (+M-effect of the C 2 H 5 O group).

The formation reactions of hemiacetals and acetals are reversible, so acetals and hemiacetals are easily hydrolyzed by excess water in an acidic environment. Hemiacetals are stable in an alkaline environment, since the alkoxydione is a more difficult leaving group than the hydroxide ion.
The formation of acetals is often used as a temporary protection for the aldehyde group.
Connecting water. Addition of water to a carbonyl group - hydration- reversible reaction. The degree of hydration of an aldehyde or ketone in an aqueous solution depends on the structure of the substrate.
The hydration product, as a rule, cannot be isolated in free form by distillation, since it decomposes into its original components. Formaldehyde in an aqueous solution is more than 99.9% hydrated, acetaldehyde is approximately half, acetone is practically not hydrated.

Formaldehyde (formaldehyde) has the ability to coagulate proteins. Its 40% aqueous solution, called formaldehyde, used in medicine as a disinfectant and preservative for anatomical preparations.
Trichloroacetic aldehyde (chloral) is completely hydrated. The electron-withdrawing trichloromethyl group stabilizes chloral hydrate so much that this crystalline substance splits off water only during distillation in the presence of dehydrating substances - sulfuric acid, etc.

The basis of the pharmacological effect of chloral hydrate CC1 h CH(OH)2 lies the specific effect on the body of the aldehyde group, which determines the disinfecting properties. Halogen atoms enhance its effect, and hydration of the carbonyl group reduces the toxicity of the substance as a whole.
Addition of amines and their derivatives. Amines and other nitrogen-containing compounds of the general formula NH 2 X (X = R, NHR) react with aldehydes and ketones in two stages. First, nucleophilic addition products are formed, which then, due to instability, eliminate water. In this regard, this process is generally classified as a reaction attachment-elimination.

In the case of primary amines, substituted imines(they are also called Schiff's bases).
Imines are intermediate products of many enzymatic processes. The production of imines goes through the stage of formation of amino alcohols, which are relatively stable, for example, when formaldehyde reacts with α-amino acids (see 12.1.4).
Imines are intermediate products in the preparation of amines from aldehydes and ketones by reductive amination. This general method involves reducing a mixture of a carbonyl compound with ammonia (or an amine). The process proceeds according to the addition-elimination scheme with the formation of an imine, which is then reduced to an amine.
When aldehydes and ketones react with hydrazine derivatives, they produce hydrazones. This reaction can be used to isolate aldehydes and ketones from mixtures and identify them chromatographically.

Schiff bases and other similar compounds are easily hydrolyzed by aqueous solutions of mineral acids to form the starting products.
In most cases, reactions of aldehydes and ketones with nitrogenous bases require acid catalysis, which accelerates the dehydration of the addition product. However, if the acidity of the medium is increased too much, the reaction will slow down as a result of the conversion of the nitrogenous base into the non-reactive conjugate acid XNH 3+.
Polymerization reactions. These reactions are characteristic mainly of aldehydes. When heated with mineral acids, aldehyde polymers decompose into their original products.
The formation of polymers can be considered as the result of a nucleophilic attack by the oxygen atom of one aldehyde molecule on the carbonyl carbon atom of another molecule. So, when formaldehyde stands, the polymer of formaldehyde - paraform - precipitates in the form of a white precipitate.
5.4. Condensation reactions
The presence of a CH acid center in an aldehyde or ketone molecule results in the α-hydrogen atoms of these carbonyl compounds having some proton mobility. Under the influence of bases, such protons can be eliminated to form the corresponding carbanions. Carbanions play the role of nucleophiles towards the carbonyl substrate. This makes it possible to carry out reactions in which one molecule, as a nucleophile, attaches to the carbonyl group of another molecule of a neutral carbonyl compound. Such processes belong to condensation reactions.
Condensation is a reaction that leads to the formation of a new carbon-carbon bond, and a new, more complex molecule is formed from two or more relatively simple molecules.
Thus, in an alkaline environment, two molecules of acetaldehyde form hydroxyaldehyde with twice the number of carbon atoms.
The reaction product containing hydroxyl and aldehyde groups is called aldolem(from words ald egid and alcohol ol), and the reaction itself was called aldol condensation, or aldol addition.
The mechanism of aldol condensation. When a base acts in a carbonyl compound, a proton is removed from the α-position and a carbonyl group is formed (I), in which the negative charge is delocalized with the participation of the carbonyl group.

The anion (I) is a strong nucleophile (shown in color in the next step of the mechanism) that adds to the second (non-ionized) molecule of the carbonyl compound. As a result of this interaction, a new C-C bond appears and an intermediate alkoxide ion (II) is formed. In an aqueous environment, this anion is stabilized by removing a proton from a water molecule and turns into the final product - an aldol.

The aldol addition reaction is shown using the example of propanal (the molecule that attaches to the C=O group of another molecule is highlighted in color); A similar reaction is shown using acetone as an example.

The condensation product, the aldol, is capable of eliminating water to form an α,β-unsaturated carbonyl compound. This usually occurs at elevated temperatures. In this case, the reaction as a whole is called Croton condensation.
Condensation reactions can also occur in a mixed version, using different carbonyl compounds, and one of them may not contain a CH acid center, such as formaldehyde and benzaldehyde in the following reactions:

Aldol condensation is a reversible reaction; the reverse process is called aldol cleavage(or retroaldol reaction). Both reactions occur in many biochemical processes.
5.5. Reduction and oxidation
Recoveryaldehydes and ketones are carried out using complex metal hydrides LiAlH 4, NaBH 4. The reaction involves a nucleophilic attack on the carbonyl carbon atom by a hydride ion.
Subsequent hydrolysis of the resulting alcoholate produces a primary or secondary alcohol.

OxidationThe transformation of aldehydes into carboxylic acids is carried out under the influence of most oxidizing agents, including atmospheric oxygen. Ketones do not oxidize under mild conditions.

Silver oxide in the form of an ammonia complex 2 OH (Tollens' reagent) oxidizes aldehydes into carboxylic acids, releasing metallic silver. This is where the name comes from - reaction "silver mirror"
Aldehydes are also easily oxidized by copper(II) hydroxide in an alkaline medium.
Both of these reactions are often used qualitatively to detect the aldehyde group, although they are nonspecific with respect to aldehydes: for example, polyhydric phenols, aminophenols, aromatic amines, hydroxyketones and other easily oxidized compounds are subject to oxidation by these reagents.
R-C-OR" + ROH: N
In alkaline hydrolysis, the leaving group (RO®) is | appears very bad and no reaction is possible. This property - the stability of acetals in an alkaline environment - is used when it is necessary to protect the carbonyl group. Protection of one or another functional group (in amines, alcohols, phenols, olefins, mercaptans, CH-acids, etc.) is a very important task in organic synthesis (Chapter XXII). [We illustrate this using the example of the synthesis of glycerol aldehyde from readily available acrolein.
CH2=CH-Cf° + KMnO, N
The action of potassium permanganate directly on acrolein leads to the oxidation of both ^C=CX^ and the aldehyde-C group: CH2-CH-C he he he
acrolein glyceric acid uO HC1.
This requires protection of the aldehyde group, which can be achieved by converting it to an acetal, for example, by the action of ethanol in the presence of hydrogen chloride.
3-chloropropanal
CH2-CH2-C-OS2H5
1D-diethoxy-3-xyaorpropane
The latter immediately attaches to the double bond simultaneously with the formation of an acetal. The key stage of the synthesis is the regeneration of the double C=C bond as a result of de-chlorination with alkali with the preservation of an acetal that is stable in an alkaline environment.
CH2-CH-Cr-OC2H5
OH OH H 1, 1-diethoxy-2,3-dihydroxypropane
Acid hydrolysis of the acetal under mild conditions gives the desired glyceraldehyde:
R°g^ H3Oe UR
sn2-sn-schn? sn2-sn-on he os2n5 he he n
2,3-dihydroxypropanal, glyceraldehyde Due to steric hindrance, ketones react with alcohols to form hemiketals much more difficult compared to aldehydes that form hemiacetals, especially with bulky groups in the ketone or alcohol.
To protect the carbonyl group, it is convenient to use glycols that form cyclic acetals, for example:
^O c© ^o-sn2
CH3CH2CH- + CH2-CH2 -H- CH3-CH2-C I
N OH OH N 0 СНз
2-ETHL-1,3-dioxalane
This is important primarily for ketones, which do not tend to form ketals when interacting with ordinary alcohols. The intramolecular formation of hemiacetals by oxyaldehydes and oxyketones is characteristic of carbohydrates; see Chapter XXIII for details.
Addition of carboxylic acids. Aldehydes, by analogy with alcohols, can add carboxylic acids (preferably their anhydrides), forming acylals:
acetaldehyde acetic anhydride ethylidene diacetate
Polymerization of aldehydes. Lower aldehydes (formaldehyde, worse - acetaldehyde) are capable of polyerization, the initiator of which is usually water.
Nosn2-o-sn2-on + n-s
Etc. - HO^CH2O^H
The nature of polymer products depends on the conditions? Section.
W In aqueous solutions, formaldehyde forms oligomeric linear polymers. When such a solution is evaporated, a solid product, paraformaldehyde, is formed. containing from 8 to 100 oxymethylene units. Water, initiating polymerization, simultaneously breaks down the polymer, hydrolyzing it, therefore, it is impossible to obtain a high-molecular polymer in aqueous solutions; when heated, ratformaldehyde, especially with acids, breaks down, turning into gaseous formaldehyde, |^if this happens in a closed vessel - into trioxane "."pl. 64 "C, boiling point 115°C).
trioxane
but-|-сн2о--н -?- 9^у
paraformaldehyde
The tempting idea of obtaining a high molecular weight (L > 1000) polymer from formaldehyde attracted many famous chemists. Polyformaldehyde was first described by A. M. Butlerov in the middle of the 19th century. The polymer received its rebirth thanks to the work of the German chemist G. Staudinger, one of the founders of polymer chemistry, who carried out basic fundamental research on the synthesis and properties of high-molecular polyformaldehyde, including chemical methods for increasing its stability. However, it was possible to overcome the enormous difficulties with the engineering implementation of the synthesis and establish the industrial production and processing of high-molecular polyformaldehyde for the first time only in 1959 (Dupont).
Currently, polyformaldehyde is obtained in the form of a homopolymer with terminal hydroxy groups converted into simple ones to prevent depolymerization
or esters (Delrin, Tenac), or a copolymer of formaldehyde with 2.5-3.0% ethylene oxide, 1,3-dioxolane
(I J) and others (celcon, SFD, hostaform) with molecular O
weighing 40-120 thousand.
CH3-C-O-J-CH2OJ-C-CH3
polyformaldehyde (delrin, tenac)
Polyformaldehyde, as an excellent construction material, is increasingly used in machinery, instrument making, and for spinning fibers.
79.3.1.3. Reactions with halogen-centered nucleophiles
Halogenoanions are weak nucleophiles (good leaving groups), and HHal forms unstable addition products with aldehydes and ketones, as noted above
In all the approaches to the problem of selectivity that we considered above, the “game” was based on variations that directly affected the participants in the main process: the nature of the substrate and/or reagent, the reaction conditions, or even the nature of the reaction itself changed. Although in each case it was possible to ensure the selectivity of the required transformation, sometimes this success was achieved at a high price, since it was necessary to “adjust” any of the main synthesis methods to the solution of a particular problem, in other words, using the previously used metaphor of “getting inside a black box.” In practice, in many cases . A different approach to the problem of selectivity turns out to be more advantageous. Let us explain it with the following schematic example.
Let's consider a certain substrate A-X, for which the method of converting it into the product A-Z has been well developed. Let us now assume that the specific task is the selective conversion of the substrate Y-A-X, where Z is a group similar in properties to group X, into the product Y-A-Z. You can, of course, try, for example, to modify the main reaction so that it affects only group X and does not affect group Y at all. However, such a path can turn out to be very labor-intensive, since it will be necessary to modify an already well-developed and possibly complex method, and it is possible that for each new Y in systems of type Y "-A-X this work will have to be done anew. Fortunately, there is a different principle for solving this kind of problem. The essence of the ggo is to temporarily remove the group Y from the game and thereby transform the bifunctional substrate Y-A-X into a monofunctional one, to which the usual method of transforming X into Z in its canonical form is applicable. This can be achieved by using some simple reactions that transform the function Y into a group that is inert under the conditions of the main reaction and allows a painless return from it to the original function Y at later ones. stages of synthesis.
Such masking, or protection of functions, is a technique extremely widely used in the practice of organic synthesis. It is easy to see that this removes the problem of selectivity of the main reaction, but the question arises about the selectivity of placing a protecting group on the function Z without affecting the related function X. However, in the general case, finding a solution to this problem is already incomparably easier for a number of reasons. Firstly, methods for introducing protection belong to the category of transformations of functional groups, which are relatively simple in chemistry and for which dozens of methods have been developed, which makes them applicable to almost all conceivable cases. Secondly, the structure of the protecting group can be varied within very wide limits, since it will be removed in subsequent stages, and its nature cannot affect the formation of subsequent products of the synthetic chain*. Due to these circumstances, the range of reactions that can be used to protect a given functional group is extremely wide, which reliably ensures the required selectivity of the protective group. To illustrate the application of the “protective approach” to the problem of selectivity, let us consider the restoration of the already familiar model trifunctional system 156 (Diagram 2.86).
| Scheme 2.86 |
Previously, using the same system, we showed how selective reduction of only the formyl group or the formyl icarbomethoxy groups can be achieved by varying the nature of the hydride reducing agent (see Scheme 2.73). But what if you want to selectively restore only the carbomethane group? If we consider that this function will be less active with respect to any of the conventional hydride reducing agents than the formyl group, it may seem that the required transformation cannot be carried out at all using reagents of this type. However, in fact, the situation can be easily corrected by protecting the carbonyl group by converting it into an acetal group using, for example, an acid-catalyzed reaction with ethylene glycol. Since acetals are stable to a wide variety of nucleophiles, the ester group of modified substrate 188 can be reduced using any hydride reducing agent. The resulting alcohol 189 differs from the required product 190 only in the presence of acetyl protection, but the latter is easily removed by acid-catalyzed hydrolysis. Thus, the almost intractable problem of selective reduction of carbomethoxy group in the presence of an easily reducible aldehyde function is easily solved by using a “protective approach”.
Let us now look more specifically at some methods for protecting the most important functional groups, starting with the carbonyl function.
The aietal protection mentioned above can, in principle, be applied to any carbonyl compound using a wide variety of alcohols or glycols, but the rate of this reaction, depending on the specific nature of the substrate, can vary by several orders of magnitude. This allows, in particular, to clearly differentiate the aldehyde and ketone functions, since the former is a more active electrophile and can be converted into an acetal much more easily. Let us consider as an example a specific synthetic problem in which this particular technique was effectively used.
Using the same example, it is convenient to show how reverse selectivity of reduction can be ensured. For this purpose, the aldehyde group is first protected by thioacetal protection (Scheme 2.88). Since thioacetals are quite stable under slightly acidic conditions, the resulting product 194 can be further converted into a deprotected derivative 195. A specific feature of thioacetals is their ability to quite easily undergo solvolysis when treated with mercury (or cadmium) salts. By such processing from the product 195 obtain a monosubstituted derivative 196, in which this time the keto group is protected, and the aldehyde group can be further reduced or used in any other reactions with nucleophilic reagents.
There are often cases when it is necessary to differentiate between a regular carbonyl group and the same group conjugated with a double bond. Since the presence of such conjugation significantly reduces the electrophilicity of the carbonyl center, acetalization in such polyfunctional systems will proceed with high selectivity, affecting only the isolated carbonyl function. This technique, especially often used in steroid chemistry, makes it possible at subsequent stages to use the enone group preserved in the molecule in such transformations as, for example, Michael addition.
It is convenient to consider the problems that arise when it is necessary to carry out selective protection of hydroxyl groups using examples from the chemistry of carbohydrates. Let's say that we need to selectively carry out the reaction at the primary hydroxyl group at C-6 of a-methyl-O-glucopyranoside (197) (diagram 2.89).
Obviously, to achieve this goal, it is necessary to first protect the other three hydroxyl functions present in the molecule. A possible way to solve this problem is the synthesis of triacetate 198. However, direct transformation 197 V 198 difficult to carry out, because acetylation is a low-selective reaction that occurs with primary alcohols faster than with secondary ones. Therefore, we have to resort to a workaround - the synthesis of triphenylmethyl (trityl, Tg) ether 199. The introduction of trityl protection at primary hydroxyls is easier than at secondary ones, since the reactions of the bulky trityl group are very sensitive to the spatial shielding of the attacked center. Indeed, glucoside treatment 197 trityl chloride in pyridine leads to monotrityl ether in high yield 199. In this compound, the primary hydroxyl is protected, which should be free in the target compound. This, however, should not confuse us: the main thing is that we managed to “tag” it somehow, i.e. distinguish from others. At the next stage, we need to close all other hydroxyl groups, for which it is quite possible to use the standard method of acetylation with acetic anhydride in pyridine. In the resulting derivative 200 There are two types of protective groups, which differ sharply in their properties, in particular, in stability with respect to acidic reagents. Therefore, converting this product to the target triacetate 198 can be carried out with high selectivity by hydrolysis in a slightly acidic environment.
| Scheme 2.89 |
Using the example considered, it is instructive to trace some general principles for the use of protecting groups. The selectivity of the final result in the shown sequence of transformations is achieved, on the one hand, by the selectivity of introducing the first protection, due to both its properties and the properties of the protected function, and on the other hand, by the selectivity of removing one of the protections, due only to differences in the properties of these groups as such. Thus, the selectivity of protection and the selectivity of deprotection are controlled by completely different factors and therefore constitute two powerful and independent ways of controlling the selectivity of the entire synthesis.
The problem of selective protection of the hydroxyl group arises extremely often in total synthesis. That is why a very sophisticated system of protection has been created for the alcohol function literally “for all occasions.” Some of the most commonly used protections are shown in Diagram 2.90. All the derivatives shown are among the generally quite common products of transformation of the hydroxyl group: these are esters (201-203), acetals (204, 205), ethers (206-209) and silyl ethers (210, 211) . The preparation of all these derivatives is carried out according to the general scheme of electrophilic substitution of the hydrogen of the hydroxyl group, however, the methods for introducing specific protections vary greatly and cover both the acidic, neutral, and alkaline regions. The ease of the reaction of establishing one or another protection depends on the nature of the alcohol hydroxyl, i.e., on the structural features of the fragment containing the hydroxyl substituent. So, for example, the relative reactivity of alcohols in such reactions can be represented by the series: “-AlkOH > v/ao/>-A1UN > tert-MkOI; equatorial ROH > axial ROH. By exploiting differences in the reactivity of alcohol functions, it is possible to differentiate these groups quite subtly by selectively introducing suitable protections.
The range of conditions under which the protection of alcohol hydroxyls is stable covers almost the entire region in which the main reactions used in organic synthesis can be carried out (except for superacid media). In general, ethers, acetals and ketals are characterized by high stability towards bases and nucleophiles, as well as oxidizing and reducing agents; for esters - to electrophiles and oxidizing agents and, in a fairly wide range, to acids; for silyl ethers - to oxidizing and reducing agents and electrophiles of some types. Therefore, to ensure the safety of the alcohol group under the conditions of almost any reaction that occurs with the participation of other available functions, you can always select some kind of protection from the available rich set of options.
| Scheme 2.90 |
The conditions for removing the listed protections are also very diverse: these are acidic or alkaline solvolysis, catalytic hydrogenolysis, reduction with complex hydrides or alkali metals in liquid ammonia and cleavage under the influence of specific reagents such as, for example, unsolvated fluoride ion (for silyl derivatives) or trimethyliodosilane ( for methyl esters, stable to most other reagents). Within each type of protection there are subtle gradations of resistance in relation to the conditions under which they are removed. For example, in the group of esters, resistance to alkaline solvolysis increases in the series: ChCCOO-R< C1CH 2 COO-R < CH 3 COO-R < C 6 H 5 COO-R < QHsNHCOO-R. Аналогично изменяется стабильность силиловых эфиров в условиях сольволиза в ряду: Me 3 Si-O-R < Me 3 CSi(Me 2)-О-R < МезС81(Рп 2)-О-R. Очень важной является возможность удаления силиль-ной группы при действии фторид-иона, что позволяет снимать эту группу, не затрагивая какие-либо другие защиты. В группе простых эфиров резко различными будут условия снятия защит при замене алкильной группы на ал-лильную, бензильную или тритильную. Так, удобным методом снятия ал-лильной защиты является двустадийная процедура: изомеризация в пропе-ниловый эфир под действием /я/>potassium e/r-butylag in absolute DMSO (or under the action of rhodium complexes) and hydrolysis under slightly acidic conditions (see Scheme 2.90). The benzyl group can be removed either under neutral conditions by hydrogenolysis over a palladium catalyst or by one-electron reduction with sodium in liquid ammonia. Tritap and its close analog p-methoxytrityl protection are very similar in their properties, but they differ so much in the rate of acid solvolysis that removing the p-methoxytrityl group while preserving the trityl group is not a particular problem.
The variety of methods for protecting the hydroxyl function, as well as methods for removing protecting groups, is a powerful tool that greatly facilitates the solution of all kinds of synthetic problems, one way or another related to the use of alcohol functions. Among them there may be not only tasks associated with the selective production of certain derivatives in a series of polyhydroxyl compounds, such as, for example, shown in Scheme 2.89. In a complete synthesis, it is very important to use a protection system configured in such a way as to make it possible to use a polyfunctional precursor as a substrate in a sequence of controlled transformations affecting these functions one after another.
A clear example of the success of this approach - an approach that is strategic in its meaning - is the synthesis of the biologically active natural diterpenoid zoopathenol (212), carried out by Nikolaou et al. . Retrosynthetic analysis of this structure suggested disassembly at bonds a, b, and c, which made it possible to select bromoketone 213 and triol 214 as the main synthetic blocks (Scheme 2.91). The formal route for the synthesis of the target product from these initial ones, including a sequence of a number of transformations, is also shown in Scheme 2.91 (asterisks indicate those centers in the reactants that participate in the formation of bonds at each stage).
From the point of view of the overall strategy, this plan looks quite convincing, since it involves relatively few steps, each of which involves the use of well-known reactions. However, even with a superficial analysis, it becomes clear that it is simply impossible to implement it in the presented form due to practically insurmountable obstacles caused by the multifunctional nature of all the shown reactants 213-218 in this hypothetical sequence. So, for example, although it is technically possible to imagine the formation of a C-C bond when 215 is assembled from the precursors 213 and 214 according to the Grignard reaction scheme between the aldehyde obtained by oxidation of 214 and the organomagnesium compound prepared from bromide 213, it is impossible to directly oxidize 214 to the aldehyde the required structure, as well as obtaining a Grignard reagent from 213 (due to the presence of a carbonyl electrophile in this molecule). It is easy to see that the implementation of other stages of the sequence shown is equally impossible in reality, despite the presence of well-developed methods for carrying out these transformations.
| Scheme 2.91 |
Obviously, it would be absolutely pointless to try to implement at least one of the stages of this plan with substrates 213-218. However, in fact, the synthesis of 212 was successfully carried out in full accordance with the plan shown above and using compounds 213 and 214 as starting materials, which, however, were included in the synthetic chain in the form of protected derivatives (see Scheme 2.92).
The synthetic equivalent of triol 214 was derivative 219, in which all three hydroxy groups are protected differently. Selective removal of the tetrashdropyranyl protection liberates the desired primary hydroxyl, which is further oxidized to the desired aldehyde 220. As noted, ketobromide 213 cannot be directly used to prepare the corresponding Grignard reagent. However, there is nothing to prevent the conversion of 213 to the corresponding ketal, from which the desired reagent 221 can be easily obtained. The reaction of 220 with 221, subsequent oxidation of the single unprotected hydroxyl group of product 222, and a repeat Grignard reaction on the resulting carbonyl group present no problem. Product 223 contains two double bonds, but only one of them must be converted into the epoxide required for subsequent construction of the oxepane ring. For the epoxidation of 223, one cannot use the most commonly used reagents for this purpose, such as peracids, because they will primarily attack the more nucleophilic trisubstituted double bond. In order to provide the required oxidation selectivity, the silyl protection was removed (by the action of an unsolvated fluorine anion), and the resulting allylic alcohol was further oxidized with tert-ВuUN - reagent for selective epoxidation of double bonds in allylic alcohols. The key stage of the entire synthesis, the intramolecular cyclization of epoxide 224 with the formation of a seven-membered ring, proceeds quite selectively, since the secondary hydroxyl, the most dangerous competitor of the reacting tertiary hydroxyl group, is reliably protected. The cyclization product diol 225 was further converted to ketone 226 via standard oxidation of the 1,2-diol moiety, after which only a few rather trivial transformations were necessary to complete the synthesis of 212.
| Scheme 2.92 |
It is obvious that the success of the entire synthesis was determined primarily by a carefully thought-out choice of the system of protecting groups in the starting compounds. Indeed, the presence of three different protecting groups in 219, a derivative of the original triol 214, made it possible to remove each of them precisely at the moment when it was necessary to carry out one or another transformation selectively involving a specific hydroxyl function, and placing protection on the ketone function in bromide 213 ensured safety ketone fragment throughout the synthetic sequence. It is noteworthy that in the synthesis of this multifunctional target structure, manipulations with protective groups were minimized and the inclusion of auxiliary operations of setting and removing additional protections at any stages was not required.
Until now, we have talked about protected compounds as derivatives that ensure the preservation of a particular function under conditions of synthetic transformations. However, often the same group can serve as protective in one series of reactions and functional in another. Some examples illustrating the importance of this aspect of the use of protecting groups in synthesis will be discussed below.
Perhaps the simplest and most obvious is the case with ester protection of the alcohol group. As we noted above, this protection allows the alcohol function to be preserved under conditions of reactions such as oxidation or glycosylation. However, no less important synthetically is the ability of esters, especially such as trifluoroacetates or triflates, to serve as active electrophiles in reactions with carbanionic nucleophiles to form a C-C bond (see, for example, Scheme 2.79).
Another classic way to protect alcohols is to convert them into trityl ethers. Most often, this method is used to exclude the possibility of electrophilic substitution of hydrogen in the corresponding hydroxyl group. However, in the case of secondary alcohols, the transition to trityl groups significantly facilitates the separation of the hydride ion from the a-CH fragment under the action of specific catalysts such as the trityl cation, as a result of which disproportionation can quite easily occur with the formation of a ketone fragment and triphenylmethane . Scheme 2.93 shows an example of using this feature of trityl protection to carry out selective oxidation of a secondary alcohol group in a bifunctional substrate 227 .
| Scheme 2.93 |
It is well known that the transformation of an aldehyde carbonyl into a dithioacetal function ensures the safety of this carbonyl under conditions of nucleophilic addition, oxidation or hydride reduction reactions. But no less important for the synthesis is the fact that dithioacetals can serve as convenient precursors for the generation of the corresponding carbanion reagents (under the action of bases such as butyllithium), and in the next section we will take a closer look at the specifics of this application of dithioacetals.
The conversion of ketones into ketals is a traditional method of protecting this fragment under reduction conditions, especially useful in cases where selective protection is possible at one of the carbonyl groups of the substrate. Thus, monoketal 228 (Scheme 2.94) can be easily and selectively obtained from the corresponding diketone, since the second ketone group (at C-17) in this compound is sterically hindered. Reduction of 228 with sodium borohydride gives (after hydrolysis of the protective group) ketoalcohol 229 in almost quantitative yield - the result can be said to be banal. However, it turns out that when reducing the same substrate 228, reverse regioselectivity can be ensured with the same completeness, namely, exclusive reduction at the C-3 center. This paradoxical, at first glance, result is achieved if reduction is carried out using diiodosilane, a reagent for the specific cleavage and hydrogenolysis of the dioxolane group. Thus, in the reaction 228 → 230, the ketal group (just a disguised equivalent of the keto group!) acts as a function with rather unusual properties.
| Scheme 2.94 |
Among acid derivatives, amides occupy a special place due to their reduced electrophilicity and, accordingly, increased stability under the conditions of methods usually used to break down other carboxyl derivatives. In general, however, amide protection is not used very often in synthesis precisely because of the stringency of the conditions required for the regeneration of the carboxyl function (see examples in the work). Nevertheless, it was with the use of amides that it was possible to significantly simplify the solution to the problems of selectivity in the Michael reaction in the series of derivatives of a,p-unsaturated acids. Thus, it is known that the interaction of esters of such acids with organomagnesium or lithium compounds usually leads to the formation of mixtures of 1,2- and 1,4-addition products. In some (but not all!) cases, the problem of selective production of 1,4-adducts can be solved using cuprate reagents. The situation is greatly simplified if we take dimethylamides like 231 (see diagram 2.95) as Michael acceptors. Due to the presence of the dimethylamide fragment, the attack of the nucleophile on the carbonyl carbon atom is completely blocked, and reactions with organolithium reagents of various natures proceed exclusively as a 1,4-addition. Moreover, the carbanion intermediate formed at the first stage is sufficiently stable under Michael addition conditions, which makes it possible to further introduce it into reactions with a wide range of electrophiles and thus obtain a variety of addition products of C-nucleophiles and C-electrophiles at the double bond of the substrate type 231. The same result can be achieved when working with trimethylhydrazides of acids, such as 232 .
| Scheme 2.95 |
This section has outlined some general principles for the use of protecting groups, with examples relating to the chemistry of alcohol and, to a lesser extent, carbonyl groups. To date, a very sophisticated system of protection for almost all main functional groups has been developed, and intensive research in this area continues. Thus, in the first edition of the monograph on protective groups (Green, Protective Groups in Chemistry, 1981) describes approximately 500 different protections for five types of functional groups. By the time the second edition of this monograph was published in 1991)